Printed from acutecaretesting.org
April 2017
Citrate anticoagulation during CRRT for acute kidney injury




SummaryThis article discusses the use of citrate anticoagulation during continuous renal replacement therapies (CRRTs) which are prescribed for patients with severe acute kidney injury (AKI). Where heparin was traditionally the anticoagulant of choice for CRRT, regional citrate anticoagulation (RCA) has now emerged as the safest and most effective way of anticoagulating blood during CRRT.
Acute kidney Injury
Acute kidney injury (AKI), a clinical syndrome with many possible causes, is simply defined as an abrupt decrease in kidney function over a period of hours or days, evidenced by recent increase in serum creatinine concentration and/or reduction in urine output [1].
It is distinct from chronic kidney disease (CKD) in which decrease in kidney function is slowly but irreversibly progressive, occurring over a period of months and years. AKI can of course occur in those with underlying CKD, and a diagnosis of AKI is associated with greater than normal risk of an eventual diagnosis of CKD [2].
Internationally agreed guidelines [1] state that a diagnosis of AKI can be made if:
• serum creatinine increases by ≥ 0.3 mg/dL (26.5 µmol/L) within 48 hours; or
• there has been a 50 % or greater increase in serum creatinine within the past 7 days; or
• urine volume is < 0.5 mL/kg/h for 6 hours
AKI encompasses the full spectrum of severity from a mild decrease in kidney function (stage 1 AKI) to renal failure (stage 3 AKI). Staging depends on serum creatinine measurement, with or without urine output data (Table I)
|
Stage |
Serum Creatinine |
Urine output |
|
1 |
1.5-1.9 times baseline |
< 0.5 mL/kg/h |
|
2 |
2.0-2.9 times baseline |
< 0.5 mL/kg/h for |
|
3 |
3.0 times baseline |
< 0.3mL/kg/h
|
TABLE I: The three stages of AKI severity (from ref 1)
The causes of AKI are conventionally classified under three headings: prerenal; renal (intrinsic); and postrenal, emphasizing the fact that AKI can have a specific renal etiology but can also result from primary problem outside the kidney.
Any condition that has the effect of reducing blood flow to the kidney (renal perfusion) is a potential cause of prerenal AKI; these conditions include: hypovolemia due to vomiting, diarrhea, hemorrhage, burns or septic shock; and reduced cardiac output due to, for example, heart failure and liver failure (cirrhosis). The major cause of intrinsic AKI is renal tubular necrosis, resulting from renal ischemia or ingestion of nephrotoxic drugs/toxins.
Sepsis, the most common cause of AKI in the critically ill, is due in part to poorly defined intrinsic renal injury akin to renal tubular necrosis [3]. Finally, postrenal AKI results from any condition that obstructs urine flow distal to the kidney; these include: renal stones (calculi); prostate disease (tumor/hypertrophy); bladder dysfunction; and urethral stricture.
Management of AKI – renal replacement therapies for AKI
There is no specific treatment for AKI and management is largely supportive with treatment directed at the underlying cause. Withdrawal of nephrotoxic drugs, fluid resuscitation, and correction of electrolyte and acid-base disturbance are all part of this general supportive management [5]. Administration of vasopressor or inotropic drugs to restore normal blood pressure and cardiac output may be necessary.
Renal replacement therapy (RRT), the focus of this article, represents the last resort treatment for patients with severe AKI that does not respond to conservative supportive management. Around 23 % of ICU patients with AKI (i.e. around 13 % of all ICU patients) receive RRT [4].
Renal replacement therapy (RRT) is the generic term for all treatments seeking to replace kidney function. Transplantation of donor kidney to those with end stage renal disease can be considered the ultimate RRT [6], but the term is more often reserved for those therapies that replace the blood filtering and homeostatic functions of the kidney by dialysis, a process that utilizes a semi-permeable membrane as the blood filter. Two broad types of dialytic RRT are available for treatment of AKI (and for that matter, end-stage CKD): peritoneal dialysis and hemodialysis.
Hemodialysis, hemofiltration and hemodiafiltration
Modern dialysis machines have the option for hemofiltration (HF) as well as hemodialysis (HD), allowing the possibility of three different dialytic modalities for the treatment of AKI: hemodialysis (HD); hemofiltration (HF); and hemodiafiltration (HDF).At the core of all three modalities is the semi-permeable membrane that constitutes a blood filter. As blood pumped from the patient flows on one side of this membrane, excess water and waste solutes pass across the membrane. The filtrate or effluent is pumped to waste as the filtered blood is returned to the patient.
For solutes like urea and creatinine, which must be removed from blood, the concentration gradient is such that the direction of diffusion is from blood to dialysis fluid, whereas for solutes like bicarbonate, which must often be added to blood to correct the acidosis associated with severe AKI, dialysis fluid with high bicarbonate concentration relative to blood ensures that diffusion is in the opposite direction, from dialysis fluid to blood. Adjustment of dialysis fluid composition and flow rate thus allows patient specific metabolic correction.
Fluid excess is often a feature of AKI that is corrected during hemodialysis. This is achieved by ultrafiltration, which depends on the hydrostatic pressure gradient that exists across the semi-permeable membrane. The relative positive hydrostatic pressure on the blood side of the membrane effectively ”pushes” water from blood to dialysis fluid.
This ultrafiltration also facilitates passage of solute molecules, which are dragged along with water during ultrafiltration by a mechanism called solute convection (or solvent drag). The combined effect of solute diffusion, solute convection and ultrafiltration during hemodialysis aims to ensure that fluid volume and composition within both extracellular and intracellular compartment is restored, as close as is possible, to that which pertains among those with normally functioning kidneys.
Hemodiafiltration (HDF) is the combination of HF and HD. It allows optimal clearance of low-molecular-weight solutes by diffusion to dialysis fluid, combined with optimal clearance of high-molecular-weight solutes by convection and ultrafiltration. As with HF, HDF requires continuous replacement fluid; and as with HD, HDF requires dialysis fluid.
Initiation and timing of RRT for AKI – intermittent versus continuous RRT
All renal replacement therapies (PD, HD, HF and HDF) can be delivered intermittently (i.e. typically in 3-4 hourly sessions) or continuously until kidney function is restored. For patients with end stage renal failure, who require life long (chronic) RRT, the treatment is exclusively intermittent hemodialysis (IHD), almost invariably delivered in four hourly sessions, three times a week [8].Patients with AKI only have a temporary requirement for RRT to support renal function during resolution of the critical/acute illness that precipitated AKI. The mean duration of RRT among patients with AKI is around 13 days [10], although in particular cases it may only be necessary for a few days, or extend for many weeks.
Traditional (absolute) indicators for initiation of RRT in AKI include:
• severe uremia (plasma urea > 30 mmol/L)
• signs of uremic encephalopathy
• acidosis (pH < 7.25, bicarbonate < 10 mmol/L)
• acute pulmonary edema
It may be beneficial to start treatment before these extreme effects of AKI are evident, but optimal timing for initiating (and stopping) RRT is currently unclear [1] and the subject of ongoing research [11,12]. There is currently no reliable data to support a particular type of RRT for AKI and consequently all types of RRT have been employed.
Current guidelines [1], however, reflect a consensus that, whatever the modality, RRT should be delivered continuously rather than intermittently to the most critically ill (hemodynamically unstable) patients. Recent surveys [13,14] indicate that nowadays continuous renal replacement therapies (CRRTs) are used much more frequently than intermittent therapies for AKI, and that the most commonly used CRRT is continuous venous venous hemofiltration (CVVHF), followed by continuous venous venous hemodiafiltration (CVVHDF).
Anticoagulation during continuous renal replacement therapy (CRRT)
CRRT, in common with all other renal replacement therapies except peritoneal dialysis, involves pumping blood through a nonphysiological, extracorporeal circuit. The inherent property of blood to coagulate (clot) on contact with nonphysiological surfaces determines that in order to maintain a ”clot free” patient extracorporeal circuit, blood must be anticoagulated.CRRT has a particular need for anticoagulation, compared with intermittent modalities, because of the prolonged treatment sessions, and the relatively slow rate at which blood is pumped through the circuit [15]. Additionally, the blood of patients who require CRRT is often already in a relative procoagulant state due to underlying critical illness (e.g. sepsis) or trauma [16].
However, systemic heparin anticoagulation, which cannot be avoided, inevitably carries with it an increased risk of bleeding, so that heparin anticoagulation during dialytic procedures is absolutely contraindicated for those who are currently bleeding or at higher than normal risk of bleeding.
Additionally, heparin can, in a small minority (< 5 %) of patients, cause an immunemediated condition called heparin-induced thrombocytopenia (HIT) type II that is associated with massive reduction in platelet numbers and the risk of life-threatening thrombosis [17].
The potential bleeding and HIT-II risks associated with heparin use highlighted the need for an alternative anticoagulant for some patients requiring RRT. Citrate provided such an alternative.
Citrate has consequently emerged in recent years as the anticoagulant of choice for all AKI patients requiring CRRT [1,15]. Intensive care units around the world are now reporting switching from heparin to regional citrate anticoagulation for CRRT [21]. Some authorities [22] are even recommending the use of citrate rather than heparin anticoagulation for intermittent (chronic) hemodialysis (IHD).
Citrate – the mechanism of its anticoagulant action and its application in CRRT
Citrate, which has been used to anticoagulate/preserve donated blood for transfusion for well over a century [23], prevents blood from clotting through its capacity to bind (chelate) the ionized calcium (iCa) that circulates in blood plasma. This iCa is a necessary cofactor for both platelet aggregation and several key steps in the intrinsic and extrinsic coagulation pathways that are responsible for fibrin production, and thereby, fibrin clot formation.
Blood coagulation is prevented by reduction in plasma iCa concentration to around 0.35 mmol/L (normal ref range 1.15-1.30 mmol/L). This non-physiological, severely hypocalcemic state is achieved by raising plasma citrate concentration to around 3 mmol/L [24] (normal plasma citrate concentration ~0.1 mmol/L [25]).
Although there are differences in detail outlined by Morabito et al [24], all protocols for citrate anticoagulation during CRRT involve the continuous infusion of citrate-containing solution (usually either trisodium citrate or acid-citrate-dextrose (ACD) solution) to the prefilter/dialysis line, close to where it leaves the patient, either as a separate solution or combined with predilution replacement fluid. The flow rate of this infusion is adjusted to achieve a blood citrate concentration of ~3 mmol/L, and thereby a plasma iCa of around 0.35 mmol/L.
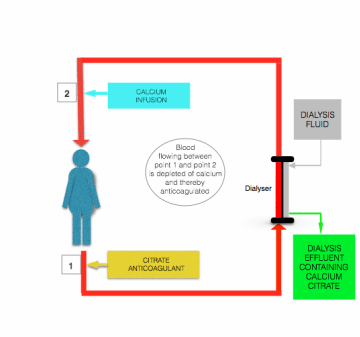
The hypocalcemia induced by citrate infusion is corrected before blood returns to the patient, by infusion of calcium-containing solution (calcium chloride or calcium gluconate) to the postfilter line close to where it enters the patient (see the figure above).
The major safety advantage of citrate anticoagulation over heparin anticoagulation is that anticoagulation is confined to the extracorporeal circuit. This so-called ”regional anticoagulation” is achieved because most of the infused citrate is removed (as either citrate anions or calcium-citrate chelate) from blood during passage through the filter/dialyzer [24].
Any remaining citrate in postfilter blood is diluted in systemic circulation and rapidly metabolized to carbon dioxide and water by the oxygen-dependant Krebs cycle in the mitochondrion of tissue cells (predominantly liver cells [24]).
Metabolism of citrate is associated with production of bicarbonate: for each 1 mmol citrate metabolized, 3 mmol bicarbonate is generated. The rapid clearance of citrate (half-life of citrate in blood is normally around 5 minutes [26]) via this principally hepatic route normally ensures the absence of systemic anticoagulation during CRRT, and thereby no additional risk of bleeding.
Potential metabolic disturbances during citrate-anticoagulated CRRT
The apparent reluctance to fully embrace citrate-anticoagulated CRRT for all AKI patients until recently has been attributed to concern regarding the acid-base and electrolyte disturbances that may occur in patients receiving the treatment [15,24].The following electrolyte and acid-base disturbances have been reported in patients receiving citrate-anticoagulated CRRT [24]:
• hypocalcemia/hypercalcemia
• hypomagnesia/hypermagnesia
• hyponatremia/hypernatremia
• metabolic alkalosis
• metabolic acidosis
These disturbances can arise for a number of reasons but accumulation of citrate in the peripheral circulation (citrate toxicity) can be central to most of these disturbances. Reasons why citrate may accumulate during CRRT include:
• Impaired metabolism of citrate due to liver disease and/or reduced delivery of oxygen because of inadequate perfusion (e.g. in septic/cardiogenic shock)
• Progressive reduction in membrane patency during dialysis with consequent reduced filtration of citrate and concomitant increase in citrate delivery to systemic circulation
• Operational error – accidental increased rate of citrate infusion to the extracorporeal circuit and concomitant increased delivery of citrate to systemic circulation
• Blood product transfusion – citrate preservative present in blood products adds to the citrate load in systemic circulation arising from CRRT
Irrespective of the cause, accumulation of citrate in the peripheral circulation can result in citrate chelation of circulating ionized calcium, with consequent reduced plasma ionized calcium concentration (hypocalcemia). If sufficiently severe hypocalcemia can cause cardiac arrhythmia and, ultimately, cardiac arrest [27].
Although plasma ionized calcium is reduced during citrate accumulation (toxicity), total calcium is increased (indicating hypercalcemia) because the calcium bound to citrate is included in measured total calcium. An increase in the ratio of total to ionized calcium (normally 2.0) to > 2.1 has been found to be the most reliable signal of citrate toxicity in the absence of routinely available plasma citrate estimation [24].
The risk of increased plasma sodium (hypernatremia) associated with citrate anticoagulation is due to the high sodium content (hypertonicity) of some citrate solutions used in CRRT. For example, the 4 % solution of trisodium citrate that is used in some protocols contains sodium at a concentration of 420 mmol/L. In practice, the use of hypotonic dialysis/replacement fluids can mitigate this risk and hypernatremia is by all accounts a rare complication of citrate anticoagulation [24].
The risk of hypomagnesia (reduced serum magnesium) during citrate anticoagulation reflects the fact that, in common with calcium, magnesium is a divalent ion that can also bind to (chelate) citrate. If magnesium losses due to the chelation action of citrate are not fully compensated for by magnesium in dialysis/replacement fluids, hypomagnesia can occur [29].
Biochemical monitoring of patients receiving citrate-anticoagulated CRRT
Point-of-care testing using a blood gas analyzer sited within the intensive care unit allows rapid and convenient measurement of most parameters (ionized calcium, pH, bicarbonate, base excess, sodium and lactate) but serum total calcium and magnesium estimation are not usually available on these platforms and samples must be sent to the laboratory for these measurements.
Two recent studies [30,31] provide evidence that some blood gas analyzers may not be reliable in measuring ionized calcium at the extremely low, non-physiological concentrations (~0.3 mmol/L) present in postfilter blood, although their ability to accurately measure plasma ionized calcium in systemic circulation (where the concentration is much higher) is not in doubt.
|
Parameter measured |
Monitoring interval |
Target values or approx. reference ranges |
Purpose of measurement |
Corrective action if outside target |
|
Postfilter ionized calcium (iCa) concentration, i.e. blood sampled from postfilter port. |
Within 1 hour of start and after any citrate dose change, then every 4-6 hours |
0.25-0.35 mmol/L |
|
Adjustment of citrate dose: increase if iCa > 0.35, decrease if iCa < 0.25 |
|
Systemic ionized calcium (iCa), i.e. blood sampled from patient |
Baseline (before starting), then 1 hour after starting, then at least every 4-6 hours |
1.15-1.30 mmol/L |
|
Adjustment of calcium infusion rate; increase if iCa < 1.15; decrease if iCa > 1.30 |
|
Systemic total calcium (TotCa), i.e. blood sampled from patient
(TotCa = iCa + calcium bound to albumin + calcium bound to citrate) |
At least every 12-24 hours (must be simultaneous to systemic ionized calcium estimation) |
|
Only required for determination of totCa:iCa ratio (see below) |
|
|
Ratio of total to ionized calcium (TotCa:iCa) |
At least every 12-24 hours |
< 2.1 |
To detect citrate accumulation/toxicity sometimes called ”citrate lock” |
Options include:
|
|
Arterial blood gas analysis
|
Baseline (before starting) . Within 1 hour of starting, then at least every 4-6 hours. |
pH 7.35-7.45 bicarb 23-28 mmol/L BE –2-+3 mmol/L |
To detect acid-base disturbances: metabolic alkalosis metabolic acidosis |
Options in the case of metabolic alkalosis include:
|
|
Serum sodium |
Once daily |
135-145 mmol/L |
To detect hypernatremia/hyponatremia |
Rare occurrence – verify correct dialysis/replacement fluid |
|
Serum lactate |
Baseline (before starting), then according to clinical needs |
< 2.0 mmol/L |
To help identify those who are at risk of citrate toxicity |
Consider, along with TotCa:iCa ratio, risk of citrate toxicity |
|
Serum magnesium |
At least once daily |
|
To detect hypomagnesia |
Correction with magnesium infusion |
References+ View more
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Working Group. KDIGO clinical practice guideline for acute kidney injury. Kidney International Supplements 2012; 2: 1-138.
- Belayev L, Palevsky P. The link between AKI and CKD. Curr Opin Nephrol Hypertens 2014; 23:149-54.
- Alobadi R, Basu R, Goldstein S et al. Sepsis-associated acute kidney injury. Seminars Nephrology 2015; 35: 2-11.
- Host E, Bagshaw S, Bellomo R et al. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPD study. Intensive Care Medicine 2015; 41: 1411-23.
- Rahman M, Shad F, Smith M. Acute kidney injury: a guide to diagnosis and management. Am Fam Physician 2012; 86: 631-39.
- Pesavento T. Kidney transplantation in the context of renal replacement therapy. Clin J Am Soc Nephrol 2009; 4: 2035-39.
- Ansari N. Peritoneal dialysis in renal replacement therapy for patients with acute kidney injury. International J Nephrology 2011; Article ID 739794.
- Himmelfarb J, Ikzler T. Hemodialysis. New Eng J Med 2010; 363:1833-45.
- Sam R. Hemodialysis: Diffusion and ultrafiltration. Austin J Nephrol Hypertens 2014; 1: 1010.
- Palevsky P, Zhang J, O’Connor P et al. Intensity of renal support in critically ill patients with acute kidney injury. New Eng J Med 2008 359: 7-20.
- Negi S, Koreeda D, Kobayashi S et al. Renal replacement therapy for acute kidney injury. Renal Replacement Therapy 2016; 2: 31.
- Lim C, Tan C, Kaushik M et al. Initiating acute dialysis at earlier Acute Kidney Injury Network stage in critically ill patients without traditional indications does not improve outcome: a prospective cohort study. Nephrology 2015; 20: 148-15.
- Jamal JA, Mat-Nor MB, Mohammed-Nor FS et al. A national survey of renal replacement therapy prescribing practice for acute kidney injury in Malaysian intensive care units. Nephrology 2014; 19: 507-12.
- Jones S, Devonald M. How acute kidney injury is investigated and managed in UK intensive care units – a survey of current practice. Nephrol Dial Transplant 2013; 28: 1186-90.
- Gutierrez-Bernays D, Otswald M, Anstey C et al. Transition from heparin to citrate anticoagulation for continuous renal replacement therapy: safety efficiency and Cost. Therapeutic Apheresis and Dialysis 2016; 20: 53-59.
- Muller M, Juffermans N. Risk estimation and management of coagulopathy in ICU patients in need of invasive procedures. Netherlands Journal of Critical Care 2015; 24: 6-11.
- Chan K, Cheung Y, Chau K. Heparin-induced thrombocytopenia due to heparin lock in a hemodialysis patient: a case report. Hemodialysis International 2014; 18: 522-61.
- Pinnick R, Weigman T, Diedrich D. Regional citrate anticoagulation for hemodialysis in the patient at high risk of bleeding. New Eng J Med 1983 308: 258-61.
- Liu C, Mao Z, Kang H et al. Regional citrate versus heparin anticoagulation for continuous renal replacement therapy in the critically ill patients: a meta-analysis with trial sequential analysis of randomized controlled trials. Crit Care 2016; 20: 144.
- Bai M, Zhou M, He L et al. Citrate versus heparin anticoagulation for continuous renal replacement therapy: an updated meta-analysis of RCTs Intensive care Medicine 2015; 41: 2098-10.
- Trumper C. Citrate anticoagulation in the ICU: the Leeds experience. Br J Nurs-ing 2016; 25: 902-06.
- Buturovic-Ponikvar J. Is regional citrate anticoagulation the future of hemodialysis? Therapeutic Apheresis and Dialysis 2016 20: 234-39.
- Mollinson P. The introduction of citrate as an anticoagulant for transfusion and of glucose as a red cell preservative. Br J Hematol 2000; 108:13-18.
- Morabito S, Pistolesi V, Tritapepe L et al. Regional citrate anticoagulation for RRTs in critically ill patients with AKI. Clin J Am Soc Nephrol 2014; 9: 2173-88.
- Kutsogiannis D, Mayers I, Chin W et al. Regional citrate anticoagulation in continuous venovenous hemodiafiltration. Am J Kidney Dis 2000; 35: 802-11.
- Davenport A. Anticoagulation options for pediatric hemodialysis. Hemodialysis International 2003; 7: 168-76.
- Zaloga G. Hypocalcemia in critically ill patients. Crit Care Med 2007; 33: 1563-70.
- Mongera S, Scholle C, Voss G et al. Metabolic complications during regional citrate anticoagulation in continuous venovenous haemodiafiltration. Nephron Clin Prac 2004; 97: c131-36.
- Zakharchenko M, Leden P, Rulisek J et al. Ionised magnesium and regional citrate anticoagulation for continuous renal replacement therapy. Blood Purification 2016; 41: 41-47.
- Schwarzer P, Kuhn S-O, Stracke S et al. Discrepant post-filter ionized calcium concentration by common blood gas analyzers in CRRT using citrate anticoagulation. Crit Care 2015; 19: 321.
- O’Orazio P, Visnich H, Balasubramanian S. Accuracy of commercial blood gas analyzers for monitoring ionized calcium at low concentrations. Clin Chim Acta 2016; 461: 34-40.
References
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Working Group. KDIGO clinical practice guideline for acute kidney injury. Kidney International Supplements 2012; 2: 1-138.
- Belayev L, Palevsky P. The link between AKI and CKD. Curr Opin Nephrol Hypertens 2014; 23:149-54.
- Alobadi R, Basu R, Goldstein S et al. Sepsis-associated acute kidney injury. Seminars Nephrology 2015; 35: 2-11.
- Host E, Bagshaw S, Bellomo R et al. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPD study. Intensive Care Medicine 2015; 41: 1411-23.
- Rahman M, Shad F, Smith M. Acute kidney injury: a guide to diagnosis and management. Am Fam Physician 2012; 86: 631-39.
- Pesavento T. Kidney transplantation in the context of renal replacement therapy. Clin J Am Soc Nephrol 2009; 4: 2035-39.
- Ansari N. Peritoneal dialysis in renal replacement therapy for patients with acute kidney injury. International J Nephrology 2011; Article ID 739794.
- Himmelfarb J, Ikzler T. Hemodialysis. New Eng J Med 2010; 363:1833-45.
- Sam R. Hemodialysis: Diffusion and ultrafiltration. Austin J Nephrol Hypertens 2014; 1: 1010.
- Palevsky P, Zhang J, O’Connor P et al. Intensity of renal support in critically ill patients with acute kidney injury. New Eng J Med 2008 359: 7-20.
- Negi S, Koreeda D, Kobayashi S et al. Renal replacement therapy for acute kidney injury. Renal Replacement Therapy 2016; 2: 31.
- Lim C, Tan C, Kaushik M et al. Initiating acute dialysis at earlier Acute Kidney Injury Network stage in critically ill patients without traditional indications does not improve outcome: a prospective cohort study. Nephrology 2015; 20: 148-15.
- Jamal JA, Mat-Nor MB, Mohammed-Nor FS et al. A national survey of renal replacement therapy prescribing practice for acute kidney injury in Malaysian intensive care units. Nephrology 2014; 19: 507-12.
- Jones S, Devonald M. How acute kidney injury is investigated and managed in UK intensive care units – a survey of current practice. Nephrol Dial Transplant 2013; 28: 1186-90.
- Gutierrez-Bernays D, Otswald M, Anstey C et al. Transition from heparin to citrate anticoagulation for continuous renal replacement therapy: safety efficiency and Cost. Therapeutic Apheresis and Dialysis 2016; 20: 53-59.
- Muller M, Juffermans N. Risk estimation and management of coagulopathy in ICU patients in need of invasive procedures. Netherlands Journal of Critical Care 2015; 24: 6-11.
- Chan K, Cheung Y, Chau K. Heparin-induced thrombocytopenia due to heparin lock in a hemodialysis patient: a case report. Hemodialysis International 2014; 18: 522-61.
- Pinnick R, Weigman T, Diedrich D. Regional citrate anticoagulation for hemodialysis in the patient at high risk of bleeding. New Eng J Med 1983 308: 258-61.
- Liu C, Mao Z, Kang H et al. Regional citrate versus heparin anticoagulation for continuous renal replacement therapy in the critically ill patients: a meta-analysis with trial sequential analysis of randomized controlled trials. Crit Care 2016; 20: 144.
- Bai M, Zhou M, He L et al. Citrate versus heparin anticoagulation for continuous renal replacement therapy: an updated meta-analysis of RCTs Intensive care Medicine 2015; 41: 2098-10.
- Trumper C. Citrate anticoagulation in the ICU: the Leeds experience. Br J Nurs-ing 2016; 25: 902-06.
- Buturovic-Ponikvar J. Is regional citrate anticoagulation the future of hemodialysis? Therapeutic Apheresis and Dialysis 2016 20: 234-39.
- Mollinson P. The introduction of citrate as an anticoagulant for transfusion and of glucose as a red cell preservative. Br J Hematol 2000; 108:13-18.
- Morabito S, Pistolesi V, Tritapepe L et al. Regional citrate anticoagulation for RRTs in critically ill patients with AKI. Clin J Am Soc Nephrol 2014; 9: 2173-88.
- Kutsogiannis D, Mayers I, Chin W et al. Regional citrate anticoagulation in continuous venovenous hemodiafiltration. Am J Kidney Dis 2000; 35: 802-11.
- Davenport A. Anticoagulation options for pediatric hemodialysis. Hemodialysis International 2003; 7: 168-76.
- Zaloga G. Hypocalcemia in critically ill patients. Crit Care Med 2007; 33: 1563-70.
- Mongera S, Scholle C, Voss G et al. Metabolic complications during regional citrate anticoagulation in continuous venovenous haemodiafiltration. Nephron Clin Prac 2004; 97: c131-36.
- Zakharchenko M, Leden P, Rulisek J et al. Ionised magnesium and regional citrate anticoagulation for continuous renal replacement therapy. Blood Purification 2016; 41: 41-47.
- Schwarzer P, Kuhn S-O, Stracke S et al. Discrepant post-filter ionized calcium concentration by common blood gas analyzers in CRRT using citrate anticoagulation. Crit Care 2015; 19: 321.
- O’Orazio P, Visnich H, Balasubramanian S. Accuracy of commercial blood gas analyzers for monitoring ionized calcium at low concentrations. Clin Chim Acta 2016; 461: 34-40.
May contain information that is not supported by performance and intended use claims of Radiometer's products. See also Legal info.


Acute care testing handbook
Get the acute care testing handbook
Your practical guide to critical parameters in acute care testing.
Download now