Printed from acutecaretesting.org
October 2006
Methemoglobin



NORMAL PHYSIOLOGY
The principal function of hemoglobin, delivery of oxygen from lungs to tissue cells, depends on the variable affinity that hemoglobin has for oxygen.
This affinity is principally dependent on the local partial pressure of oxygen (pO2), but pH, partial pressure of carbon dioxide (pCO2) and concentration of organic phosphates are also significant.
Local conditions in the lungs (relatively high pO2, low pCO2, etc) are associated with high affinity, so that hemoglobin readily binds oxygen here; the product of this binding is oxyhemoglobin.
By contrast in the microvasculature of the tissues, local conditions (relatively low pO2, high pCO2, etc) are associated with low hemoglobin affinity for oxygen and oxyhemoglobin readily dissociates, releasing oxygen to tissue cells.
Heme iron – the site of oxygen binding
The adult hemoglobin molecule (HbA) comprises four folded polypeptide chains (two alpha and two beta), each of which has a porphyrin heme group attached [1]. At the center of each of the four heme groups is an atom of iron in the ferrous (Fe2+) state.
These four iron atoms are the functional centers of the hemoglobin molecule because it is here that oxygen reversibly binds to form oxyhemoglobin.
Oxyhemoglobin is a superoxo-ferriheme (Fe3+O2–) in which there is temporary partial transfer of an electron (negative charge) from the iron in heme to oxygen [2].
When oxygen is unloaded from oxyhemoglobin in the tissues, the temporarily shared electron is recaptured by the iron atom, returning to its ferrous (Fe2+) state.
Whatever the precise detail of oxygen binding to hemoglobin, it is clear that for binding to occur, the iron atoms present in each of the four heme groups must be in the ferrous state.
The only difference between hemoglobin and methemoglobin is that one or more of the four iron atoms in the methemoglobin molecule are in the ferric (Fe3+) rather than the ferrous (Fe2+) state and are therefore incapable of binding oxygen [3].
Conversion of iron from the ferrous to the ferric state represents the loss of an electron, i.e. it is an oxidative process.
The formation of methemoglobin from hemoglobin within red cells is an ongoing oxidative process that results from exposure of hemoglobin to a variety of highly reactive molecules (oxygen free radicals), produced during normal cell metabolism [4].
The effect of free-radical-mediated oxidation is not confined to the hemoglobin molecule; many molecular species in cells throughout the body are affected. If left unchecked, such oxidative molecular changes can affect function and may ultimately cause cell disruption and injury.
The red cell and its contents (including hemoglobin) are considered particularly susceptible to this oxidative stress because of the relatively high oxygen concentration present and the resulting production of oxygen free radicals [4].
Methemoglobin is also formed during unloading of oxygen from deoxyhemoglobin in the tissues if, as sometimes happens, the temporarily donated electron to form superoxo-ferriheme is not recaptured by the iron atom; this process is called auto-oxidation [5].
It has been estimated that around 3 % of hemoglobin is converted to methemoglobin daily by these two oxidative mechanisms.
Fortunately, in view of the potential threat to oxygen delivery that methemoglobin poses, there are protective mechanisms that ensure that most of this methemoglobin is converted back to hemoglobin, so that no more than 1-2 % of total hemoglobin is normally present as methemoglobin.
Physiological mechanisms for conversion of methemoglobin to hemoglobin
For methemoglobin (MHb) to be converted to hemoglobin iron in the ferric (Fe3+) state at any or all of the four heme groups, they must be reduced to the ferrous (Fe 2+) state; in other words they must gain an electron.
By far the most significant means of effecting this electron gain is an enzyme-reducing system present in red cells, known as NADH-dependent cytochrome b5-methemoglobin reductase.
Under physiological conditions this system accounts for close to 99 % of the daily methemoglobin reduction to hemoglobin.
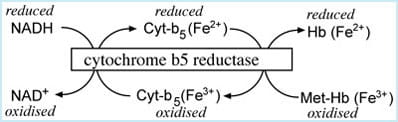
The system comprises three elements: reduced nicotinamide dinucleotide (NADH); the heme-containing protein, cytochrome b5; and the enzyme, cytochrome b5 reductase. The electron donor is NADH, a product of glucose oxidation (glycolysis).
Electrons pass from NADH to cytochrome b5 and finally to methemoglobin; the electron transfer is catalyzed by the enzyme cytochrome b5 reductase (FIGURE 1).
FIGURE 1: NADH-dependent cytochrome b5 - metHb reductase system
Another reducing pathway, which depends on the enzyme NADPH-MHb reductase, is also capable of converting methemoglobin to hemoglobin, but under normal physiological conditions this is of very minor importance.
However, this alternative pathway is significant in cases of cytochrome-b5-reductase deficiency and essential for the therapeutic action of methylene blue, the drug used to treat acquired methemoglobinemia.
Finally several general antioxidant (i.e. electron-donating) species present in red cells, such as reduced glutathione and ascorbic acid, may play a minor role in reducing methemoglobin to hemoglobin.
CAUSES OF METHEMOGLOBINEMIA
Methemoglobinemia is an abnormal increase in the concentration of methemoglobin, often expressed as an increased percentage of total hemoglobin.
Methemoglobinemia can be inherited or acquired following exposure to any one of a range of oxidant environmental chemicals and drugs.
Inherited methemoglobinemia
Several rare inherited defects in the gene that regulates the production of the enzyme cytochrome b5 reductase have been described [6,7].
The resulting congenital enzyme deficiency causes methemoglobinemia because the rate at which methemoglobin is converted to hemoglobin cannot keep pace with the rate at which it is formed. There are two main types; the most common is type I in which the enzyme deficiency is confined to red cells.
In type II, the enzyme is deficient not only in red cells but in a range of other cell types, including those of the brain [8]. This more serious but extremely rare form is associated with mental retardation and a range of neurological abnormalities that usually result in death during the childhood years.
Several inherited defects of the hemoglobin structure, all designated Hemoglobin M (HbM), result in methemoglobinemia. They are characterized by single amino acid substitutions in either the alpha or beta polypeptide chains at the region where the iron-containing heme portion is attached.
These changes have the effect of making any iron in the oxidized ferric state more stable and resistant to the effect of reducing enzyme systems than normal hemoglobin (HbA), so that methemoglobin cannot be converted to hemoglobin in those with HbM.
There is no available treatment to lower the methemoglobin caused by HbM. Those with the enzyme deficiency require lifelong treatment with an agent that has the same reducing effect as the deficient enzyme; a daily dose of ascorbic acid (vitamin C) and/or riboflavin (vitamin B2) fits this therapeutic bill.
Acquired methemoglobinemia
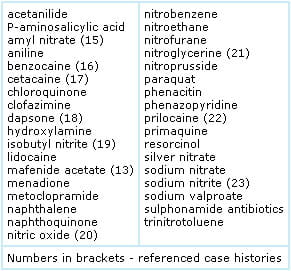
Inherited methemoglobinemia is rare; much more commonly methemoglobinemia is acquired as a result of exposure to oxidant chemicals, including a range of both prescribed and self-prescribed (over-the-counter) drugs (TABLE 1).
Acquired methemoglobinemia occurs when the rate of hemoglobin oxidation to methemoglobin – as a result of oxidant exposure – exceeds the rate at which methemoglobin can be reduced by NADH cytochrome b5-MHb reductase.
TABLE 1: Drugs or toxins than can cause methemoglobinemia
Oxidant-chemical or drug exposure can occur via ingestion, inhalation or absorption across skin or mucous membranes. Some drugs or chemicals (e.g. nitrites) have a direct oxidant effect on hemoglobin, whilst others (e.g. dapsone) owe their oxidant potential to a metabolic product.
SIGNS AND SYMPTOMS OF METHEMOGLOBINEMIA
Oxidation of iron (from the ferrous to the ferric state) in the heme portion of hemoglobin prevents oxygen binding at that site, reducing the oxygen-carrying capacity. Furthermore, oxidation of the iron at one or more of the four heme sites of the hemoglobin molecule increases affinity for oxygen at any remaining "normal" heme sites [9]. As a consequence oxygen release at tissue level is reduced.
The functional anemia that characterizes methemoglobinemia is due to the combined effect of reduced oxygen-carrying capacity and reduced release of oxygen to the tissues.
The extent and severity of symptoms are directly proportional to the percentage of hemoglobin that has been oxidized to (dysfunctional) methemoglobin. Mild methemoglobinemia (2-10 %) is generally well tolerated and, in an otherwise healthy individual, is asymptomatic.
The first sign of tissue hypoxia, evident as methemoglobin rises above 10-15 %, is cyanosis with skin taking on a classically blue/slate gray appearance.
Symptoms of more profound hypoxia, including increased heart rate, headache, dizziness and anxiety, accompany deepening cyanosis as methemoglobin rises above 20 %. Severe methemoglobinemia (>50 %) is associated with increasing breathlessness and fatigue.
Confusion, drowsiness and coma ensue; there may be seizures. Blood gas analysis reveals metabolic (lactic) acidosis consequent on anaerobic cellular metabolism. Methemoglobin >70 % is frequently fatal.
Symptoms for a given percentage of methemoglobin are generally more severe in a patient who has some pre-existing condition (e.g. anemia, respiratory or cardiovascular disease) that compromises oxygenation of tissues.
TESTS OF OXYGENATION USED IN THE CYANOTIC PATIENT
Cyanosis is an almost invariable presenting clinical sign of significant methemoglobinemia, although it is not specific for the condition.
Since cyanosis is objective evidence of inadequate tissue oxygenation, it would usually prompt the attending medical staff to order tests of oxygenation, including arterial blood gas analysis and pulse oximetry monitoring.
For the patient whose cyanosis is the result of methemoglobinemia, oxygenation indices (pO2 and sO2) measured during blood gas analysis, as well as pulse oximetry readings may remain remarkably normal, despite often profound cyanosis.
The reason for these apparently anomalous, possibly counterintuitive results [10] is worth highlighting because they can be diagnostically useful.
Partial pressure of oxygen in blood (pO2) is a parameter measured during blood gas analysis that reflects the small fraction of total oxygen that is dissolved in blood plasma, not that bound to hemoglobin.
Diffusion of oxygen from the alveoli of the lungs to blood plasma is the main determinant of pO2 and this is not impaired by methemoglobinemia, so that despite what might be severe cyanosis, pO2 remains normal in those with methemoglobinemia.
The dissociation of pO2 and cyanosis in methemoglobinemia is diagnostically useful. Administration of oxygen raises the pO2 but fails to correct cyanosis in patients with methemoglobin.
By contrast, administration of oxygen to patients whose cyanosis is the result of respiratory or heart disease also causes a rise in pO2, but in this case the rise is associated with resolution of cyanosis.
Oxygen saturation reflects the oxygen bound to hemoglobin. Since methemoglobin cannot bind oxygen, methemoglobinemia is associated with reduced oxygen saturation.
The oxygen saturation (sO2) result produced by blood gas analysis is falsely normal in patients with methemoglobinemia because it is based on a calculation that assumes a normal oxygen dissociation curve and the near absence of dyshemoglobins, neither of which of course pertain in those with methemoglobinemia.
Oxygen saturation can also be measured by pulse oximetry, a non-invasive spectrophotometric method that is based on absorbance of light at two wavelengths, 660 and 990 nm.
The computation required to calculate oxygen saturation from these absorbance measurements assumes that only oxy- and deoxyhemoglobin are present in blood; there are no dyshemoglobins. Oxygen saturation as measured by pulse oximetry is reduced in those with methemoglobinemia.
However, the reduction does not correlate with the severity of methemoglobinemia [11].
As methemoglobin increases from 2 to 30 %, oxygen saturation falls from normal (around 98 %) to around 85 %, but no further fall in oxygen saturation is seen if methemoglobin rises beyond 30-35 %. In a clinical setting this means that oxygen saturation as measured by pulse oximetry gives a falsely optimistic impression of tissue oxygenation among those with moderate to severe methemoglobinemia.
To summarize, cyanosis associated with methemoglobinemia differs from cyanosis due to other causes in two respects: it is not associated with reduced pO2 and does not respond to oxygen therapy. Oxygen saturation results obtained during arterial gas analysis are falsely normal in methemoglobinemia, and pulse oximetry readings can be misleading.
The only reliable method of measuring methemoglobin concentration and confirming a diagnosis of methemoglobinemia is CO-oximetry. Most modern blood gas analyzers have an incorporated CO-oximeter, which allows arterial blood to be spectrophotometrically examined at multiple wavelengths. All hemoglobin species have characteristic absorbance spectra.
CO-oximetry thus allows both identification and quantitation of all hemoglobin species, including methemoglobin. CO-oximetry also allows calculation of oxygen saturation; this is a more reliable method of assessing oxygen saturation in patients with methemoglobin than either pulse oximetry or blood gas analysis.
SOME ILLUSTRATIVE CASE HISTORIES
Case history 1:
Inherited methemoglobinemia is usually benign
The case [12] concerns a 25-year-old male who was sufficiently concerned about the grayish-blue color of his facial skin to seek medical advice. He had no other major complaints. On questioning he stated his exercise tolerance was good but occasionally he suffered headaches, vertigo and breathlessness. Clinical examination revealed a well-nourished and developed man with no abnormal neurological or cardiovascular signs.
The only abnormal finding was cyanosis of face, lips, ears, fingers and toes. It transpired that the abnormal skin color had been present since infancy. Past history revealed hospitalization for investigation of cyanosis at 9 months of age but no diagnosis was made at this time or during several other clinical consultations over the intervening years.
In the absence of any clinical evidence that the apparent congenital cyanosis was due to cardiac or lung disease, inherited methemoglobinemia was considered and confirmed with the finding of a methemoglobin of 40 % and absence of the red-cell enzyme cytochrome b5 reductase (Type I deficiency).
The patient was successfully treated with a daily dose of the slow-acting methemoglobin-reducing agents, ascorbic acid and riboflavin. At the 6-month follow-up methemoglobin was 20 %; cyanosis had resolved.
This case history demonstrates that the most common form of inherited methemoglobinemia, type 1 cytochrome b5 reductase deficiency, is a benign condition that can remain undiagnosed for many years.
Despite marked methemoglobinemia, the cosmetic effect of cyanosis was the only significant problem for this patient. In the words of one expert discussing the clinical effect of inherited type 1 cytochrome b5 reductase deficiency, “these patients are really more blue than sick” [6].
Case history 2:
Chocolate-brown-colored blood suggests methemoglobinemia
Blood containing a high concentration of methemoglobin is chocolate brown in color rather than the dark red of deoxygenated (venous) blood or the bright red of oxygenated (arterial) blood. As this case history [13] exemplifies, blood color is often a useful diagnostic pointer.
A 21-year-old man, JL suffered extensive burns covering 68 % body surface area following an explosion.
Surgical removal of burned skin and skin grafting were required during a prolonged period of hospitalization in a regional burns unit.
Wounds were treated with mafenide-acetate dressings to prevent infection. During the period of recovery, JL developed unexplained chronic fever, suggesting possible endocarditis (infection of the heart) and was scheduled for a diagnostic procedure called transesophageal echocardiography, which allows ultrasound examination of the heart.
The procedure involves passing an ultrasound probe down the throat to the esophagus, and in preparation for this JL’s throat was sprayed with the local anesthetic benzocaine. Within 10 minutes of the procedure JL’s condition suddenly deteriorated. Skin color turned dusky, with cyanosis around the mouth. He became increasingly lethargic.
Pulse oximetry indicated oxygen saturation was 88 % (normal 96-99 %) and he was given oxygen. Despite oxygen therapy and manual lung ventilation there was no clinical improvement or change in oxygen saturation.
A pulmonary embolus was suspected and arterial blood was sampled for blood gases. The blood sample was dark, almost chocolate brown and it was assumed that venous rather than arterial blood had been sampled, so a second sample was obtained. This, too, was similarly dark but was analyzed; blood gas results were:
| pH | 7.54 |
| pCO2 | 3.2 kPa |
| pO2 | 39.9 kPa |
| Bicarbonate | 23.6 mmol/L |
| Oxygen saturation | 98.2 % |
It remained unclear at this time what the problem was, but pulmonary embolus was excluded by raised pO2 and normal chest radiograph. The consistently dark color of arterial blood suggested a diagnosis of methemoglobinemia, which was confirmed with the finding that the methemoglobin level was 40 %.
The patient’s mafenide-acetate dressings were immediately removed and methylene blue (2 mg/kg IV) was administered, methemoglobin soon fell to 1.2 %, and JL recovered from this crisis without any long-term consequences.
After a further 2 months in hospital rehabilitating from burn injuries JL was eventually discharged home.
This case history exemplifies many aspects of acquired methemoglobinemia, including exposure to oxidant drugs (in this case benzocaine and mafenide acetate); cyanosis unresponsive to oxygen therapy; falsely normal oxygen saturation by blood gas analysis; discordance between oxygen saturation by this method compared with that obtained by pulse oximetry; and the immediate effectiveness of methylene blue therapy.
In this case it was the chocolate-brown color of arterial blood that first alerted carers to the possibility that methemoglobinemia was the problem.
Case history 3:
Newborns at increased risk of methemoglobinemia
It remains unclear why some people develop symptomatic methemoglobinemia when exposed to oxidant chemicals and drugs, whilst others do not. In the case of newborns, however, there are clear physiological reasons for susceptibility.
Newborn babies are at particular risk of methemoglobinemia for two main reasons. During fetal development and for the first 3-6 months of life, red cells contain fetal hemoglobin (HbF) rather than adult hemoglobin (HbA).
HbF is more readily oxidized to methemoglobin than HbA. In addition, the red cell enzyme cytochrome b5 reductase required to convert methemoglobin to hemoglobin is deficient during this period.
These two factors were probably significant in the case of a 6-week-old girl who was brought, in a very poor state, to the emergency room of her local hospital following a 2-day history of diarrhea [14].
On examination the baby showed signs of dehydration, and dusky skin color was noted. Vital signs included: heart rate 150; blood pressure 75/40; respirations 58/min and shallow; and rectal temperature 37.8 °C. Laboratory results included hematocrit of 25.4 %.
Severe metabolic acidosis (pH 7.12, bicarbonate 6 mmol/L and pCO2 3.2 kPa) suggested poor tissue perfusion presumed to be the result of sepsis and the baby was admitted to intensive care.
The combination of raised pO2 (28 kPa) in association with reduced oxygen saturation (91-94 %) by pulse oximetry, and dark arterial blood suggested possible methemoglobinemia.
This was confirmed with a methemoglobin of 28 %. Methylene blue was administered, and within 30 minutes the baby’s skin color retuned to normal and her general condition dramatically improved. She was discharged home 6 days after admission.
This was a case of acquired methemoglobinemia occurring without exposure to an exogenous oxidant, and it demonstrates the vulnerability of sick babies with diarrhea and acidosis to methemoglobinemia.
It is thought that nitrate-forming bacteria in the gut, or other endogenous oxidant species associated with diarrhea, may be the precipitating cause in these babies, who are already predisposed to methemoglobinemia.
SUMMARY
Methemoglobin is a useless form of hemoglobin, which is constantly being formed in the red cells of peripheral blood. However, due to the effect of an enzyme-reducing system that converts methemoglobin to functional hemoglobin, concentration remains low in health.
Increased concentration of methemoglobin, termed methemoglobinemia, reduces oxygenation of tissue cells, causing cyanosis. Severe methemoglobinemia (methemoglobin >70 % of total hemoglobin) is potentially fatal.
Blood analysis by CO-oximetry provides the means of diagnosis and methylene blue administered intravenously, the principal mode of treatment. Methemoglobinemia can be due to inherited gene defects, but much more commonly it arises as a result of exposure to oxidant drugs or environmental oxidant chemicals.
References+ View more
- Ranney HM, Sharma V. Structure and Function of Hemoglobin. In Beutler E, Lichtman M et al (eds) William's Hematology (6th edition) McGraw Hill, 2000: 345-53.
- Wittenburg J, Wittenberg B, Peisach J et al. On the state of the iron and the nature of the ligand in oxyhemoglobin. Proc Natl Acad Sci 1970; 67: 1846-53.
- Price D. Methemoglobinemia. In: Goldfrank’s toxicological emergencies (7th edition) New York: McGraw Hill 2002: 1438-39.
- Wright R, Lewader W, Woolf A. Methemoglobinemia: Etiology, pharmacology and clinical management. Ann Emerg Med 1999; 34: 646-56.
- Mansouri A, Luire A. Concise review: Methemoglobinemia. Am J Hematol. 1993; 42: 7-12.
- Jaffe E, Hultquist D. Cytochrome b5 reductase deficiency and ezymopenic hereditary methemoglobinemia. In: The Metabolic and Molecular Basis of Inherited Disease (7th edition). New York: McGraw-Hill 1995: 2267-80.
- Da-Silva S, Sajan I, Underwood J. Congenital methemoglobinemia: a rare cause of cyanosis in the newborn – a case report. Pediatrics 2003; 112e: e158-e161.
- Ywata Y, Ding L, Tanashima K et al. New variant of cytochrome b5 reductase deficiency (b5Rkurashaki) in red cells, platelets, lymphocytes and cultured fibroblasts with congenital methemoglobinemia, mental and neurological retardation and skeletal abnormalities. Am J Hematol 1992; 40: 299-305.
- Darling R, Roughton F. The effect of methemoglobin on the equilibrium between oxygen and hemoglobin. Am J Physiol 1942: 137: 56-58.
- Hammond S, Cariappa R, Eby C et al. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005; 51: 434-44.
- Watcha M, Connor M, Hing A. Pulse oximetry in methemoglobinemia. Am J Dis Child. 1989 143: 845-47.
- Hafsia R, Meddeb B, Mtimet B et al. Congenital cyanosis due to methemoglobin reductase deficiency: first reported Tunisian case. Nouv Rev Fr Haematol 1989; 31: 371-73.
- Wolak E, Byerly F, Mason T et al. Methemoglobinemia in critically ill burned patients. Am J Crit Care 2005; 14:104-08.
- Jolly B, Monico E, McDevitt B. Methemoglobinemia in an infant: Case report and review of the literature. Pediatric Emerg Care 1995; 11: 294-97.
- Modarai B, Kapadia Y, Kerins M et al. Methylene blue: a treatment for severe methaemoglobinaemia secondary to misue of amyl nitrite. Emerg Med J 2002: 19: 270-71.
- Birchem S. Benzocaine-induced methemoglobinemia during transesophageal echocardiography. JAOA 2005; 105: 381-84.
- Lunenfield E, Kane G. Methemoglobinemia: Sudden dyspnea and oxyhemoglobin desaturation after esophagoduodenoscopy. Respir Care 2004; 49: 940-42.
- Lee S, Lee J, Lee K et al. A case of methemoglobinemia after ingestion of an aphrodisiac, later proven as Dapsone. Yonsei Med J 1999; 40: 388-91.
- Jansen T, Barnung S, Mortenson C et al. Isobutyl-nitrite-induced methemoglobinemia; treatment with an exchange transfusion during hyperbaric oxygenation. Acta Anaesthesiol Scand 2003; 47:1300-01.
- Taylor M, Christian K, Patel N et al. Methemoglobinemia: Toxiciy of inhaled nitric oxide therapy. Pediatr Crit Care Med 2001; 2: 99-101.
- Bojar R, Rastegar H, Payen D et al. Methemoglobinemia from intravenous nitroglycerin: a word of caution. Ann Thor Surg 1987; 43: 332-34.
- Frey B, Keher B. Toxic methemoglobin concentration in premature infants after application of a prilocaine containing cream and peridural prilocaine. Eur J Pediatr 1999; 158: 785-88.
- Finnan A, Keenan P, O’Donavan F et al. Methaemoglobinaemia associated with sodium nitrite in three siblings. BMJ 1998; 317: 1138-39.
References
- Ranney HM, Sharma V. Structure and Function of Hemoglobin. In Beutler E, Lichtman M et al (eds) William's Hematology (6th edition) McGraw Hill, 2000: 345-53.
- Wittenburg J, Wittenberg B, Peisach J et al. On the state of the iron and the nature of the ligand in oxyhemoglobin. Proc Natl Acad Sci 1970; 67: 1846-53.
- Price D. Methemoglobinemia. In: Goldfrank’s toxicological emergencies (7th edition) New York: McGraw Hill 2002: 1438-39.
- Wright R, Lewader W, Woolf A. Methemoglobinemia: Etiology, pharmacology and clinical management. Ann Emerg Med 1999; 34: 646-56.
- Mansouri A, Luire A. Concise review: Methemoglobinemia. Am J Hematol. 1993; 42: 7-12.
- Jaffe E, Hultquist D. Cytochrome b5 reductase deficiency and ezymopenic hereditary methemoglobinemia. In: The Metabolic and Molecular Basis of Inherited Disease (7th edition). New York: McGraw-Hill 1995: 2267-80.
- Da-Silva S, Sajan I, Underwood J. Congenital methemoglobinemia: a rare cause of cyanosis in the newborn – a case report. Pediatrics 2003; 112e: e158-e161.
- Ywata Y, Ding L, Tanashima K et al. New variant of cytochrome b5 reductase deficiency (b5Rkurashaki) in red cells, platelets, lymphocytes and cultured fibroblasts with congenital methemoglobinemia, mental and neurological retardation and skeletal abnormalities. Am J Hematol 1992; 40: 299-305.
- Darling R, Roughton F. The effect of methemoglobin on the equilibrium between oxygen and hemoglobin. Am J Physiol 1942: 137: 56-58.
- Hammond S, Cariappa R, Eby C et al. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005; 51: 434-44.
- Watcha M, Connor M, Hing A. Pulse oximetry in methemoglobinemia. Am J Dis Child. 1989 143: 845-47.
- Hafsia R, Meddeb B, Mtimet B et al. Congenital cyanosis due to methemoglobin reductase deficiency: first reported Tunisian case. Nouv Rev Fr Haematol 1989; 31: 371-73.
- Wolak E, Byerly F, Mason T et al. Methemoglobinemia in critically ill burned patients. Am J Crit Care 2005; 14:104-08.
- Jolly B, Monico E, McDevitt B. Methemoglobinemia in an infant: Case report and review of the literature. Pediatric Emerg Care 1995; 11: 294-97.
- Modarai B, Kapadia Y, Kerins M et al. Methylene blue: a treatment for severe methaemoglobinaemia secondary to misue of amyl nitrite. Emerg Med J 2002: 19: 270-71.
- Birchem S. Benzocaine-induced methemoglobinemia during transesophageal echocardiography. JAOA 2005; 105: 381-84.
- Lunenfield E, Kane G. Methemoglobinemia: Sudden dyspnea and oxyhemoglobin desaturation after esophagoduodenoscopy. Respir Care 2004; 49: 940-42.
- Lee S, Lee J, Lee K et al. A case of methemoglobinemia after ingestion of an aphrodisiac, later proven as Dapsone. Yonsei Med J 1999; 40: 388-91.
- Jansen T, Barnung S, Mortenson C et al. Isobutyl-nitrite-induced methemoglobinemia; treatment with an exchange transfusion during hyperbaric oxygenation. Acta Anaesthesiol Scand 2003; 47:1300-01.
- Taylor M, Christian K, Patel N et al. Methemoglobinemia: Toxiciy of inhaled nitric oxide therapy. Pediatr Crit Care Med 2001; 2: 99-101.
- Bojar R, Rastegar H, Payen D et al. Methemoglobinemia from intravenous nitroglycerin: a word of caution. Ann Thor Surg 1987; 43: 332-34.
- Frey B, Keher B. Toxic methemoglobin concentration in premature infants after application of a prilocaine containing cream and peridural prilocaine. Eur J Pediatr 1999; 158: 785-88.
- Finnan A, Keenan P, O’Donavan F et al. Methaemoglobinaemia associated with sodium nitrite in three siblings. BMJ 1998; 317: 1138-39.
May contain information that is not supported by performance and intended use claims of Radiometer's products. See also Legal info.


Acute care testing handbook
Get the acute care testing handbook
Your practical guide to critical parameters in acute care testing.
Download now
Related webinar
Evolution of blood gas testing Part 1
Presented by Ellis Jacobs, PhD, Assoc. Professor of Pathology, NYU School of Medicine.
Watch the webinar