Printed from acutecaretesting.org
October 2005
Causes and clinical significance of increased carboxyhemoglobin




NORMAL PHYSIOLOGY
Normal cell function is crucially dependent on a continuous supply of oxygen, and a principle function of blood is delivery of oxygen in inspired air from lungs to every tissue cell.
This essential gas transport function depends on the protein hemoglobin (Hb) contained in red blood cells (erythrocytes).
Structure and function of hemoglobin
Each of the 5 × 1010 erythrocytes contained in every mL of blood contains 280 million Hb molecules. The Hb molecule comprises four polypeptide subunits (the globin portion) each of which has a heme group attached [1].
At the center of the four heme groups is an atom of iron in the ferrous state. Oxygen binds reversibly to these four iron atoms; the product is oxyhemoglobin (O2Hb).
The oxygen transport function of hemoglobin, that is its ability to pick up oxygen in the lungs, transport it around the body as O2Hb and then release it to tissue cells, is made possible by a change in the quaternary structure of the hemoglobin molecule, which alters the affinity of hemoglobin for oxygen.
The quaternary state and consequent affinity of hemoglobin for oxygen is governed principally by local partial pressure of oxygen (pO2), although pH, pCO2 and organic phosphate concentration are important modulating factors.
In the environment of the lungs, where conditions (high pO2, low pCO2) determine that hemoglobin has a relatively high affinity for oxygen, O2Hb is readily formed.
By contrast in the tissues, local conditions (low pO2, raised pCO2) result in reduced hemoglobin affinity for oxygen, thus favoring release of oxygen from hemoglobin to tissue cells. The relationship between pO2 and the relative affinity of Hb for oxygen is described in the oxygen saturation curve (Figure I).
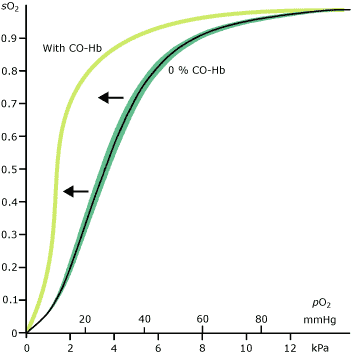 |
FIGURE I
Oxygen has to compete with other hemoglobin-binding ligands that may be present in blood for occupation of hemoglobin binding sites; among these is carbon monoxide, a colorless odorless gas produced during normal metabolism.
Endogenous production of carbon monoxide
It is more than 50 years since Sjostrand first demonstrated that carbon monoxide (CO) is produced during normal metabolism [2]. In fact, around 0.4 mL CO is produced every hour almost exclusively from the catabolism of heme-containing proteins [3].
The most abundant heme-containing protein and therefore the source of most endogenous CO is hemoglobin. At the end of their 120-day life, erythrocytes are sequestered from the circulation by the reticuloendothelial system.
Hemoglobin released from senescent erythrocytes is degraded to its constituent parts: heme and protein polypeptide. The protein is recycled but heme is metabolized further.
In a reaction catalyzed by the rate-limiting enzyme heme oxygenase, heme is converted to equimolar quantities of biliverdin, iron and CO. Biliverdin is subsequently converted to the yellow pigment bilirubin, which is excreted by the liver in bile, and iron is recycled.
Catabolism of heme derived from other heme-containing proteins, e.g. myoglobin and the cytochromes, contribute to endogenous production of CO by the same heme-oxygenase-mediated route.
There is evidence that CO is also derived from non-heme sources, e.g. lipid peroxidation [4], but compared to that derived from catabolism of heme, this is of very minor significance, indeed may only occur in pathological situations.
The biological effect of endogenous CO is due largely to the high affinity that heme has for CO and the resulting binding of CO by heme-containing proteins. By a curious quirk of nature then, heme is both the source of CO and the mediator of its biological effect.
The modulation in function of some heme-containing proteins that results from CO binding has important physiological effects.
Thus endogenously produced carbon monoxide is not, as was once supposed, simply a potentially toxic waste product of metabolism but is involved in many physiological functions, including regulation of respiration [5], neuronal signaling [6], regulation of blood pressure [7] and uterine contraction during pregnancy [8].
Of all heme-containing proteins, Hb is not only the most abundant but also exhibits the highest affinity for carbon monoxide, so that most CO in blood is bound to Hb.
Reversible binding occurs at the same iron atom on the heme site where oxygen binds; the product of this binding is carboxyhemoglobin (COHb).
This provides the means by which endogenous carbon monoxide can be transported, prior to elimination from the body by the lungs in expired air. A minimum of 0.5-1.0 % COHb is inevitably present in blood as a result of endogenously produced CO [9].
Environmental sources of carbon monoxide
In addition to the CO produced endogenously, the air we breathe contains CO, partly the result of natural processes but mostly from the incomplete combustion of hydrocarbons.
The most significant unnatural source of environmental CO is motor-vehicle exhaust. Although normally present at concentrations of less than 10 parts per million (ppm) [10], carbon monoxide in inspired air has an important additive effect on the amount of COHb in blood due to the high affinity that Hb exhibits for CO.
The combined effect of endogenous and environmental CO results in a COHb of less than 3 % for most non-smoking urban dwellers and may be just 1-2 % for those living in rural areas where air is less polluted with CO.
Cigarette smoke contains a high concentration of CO and smokers are exposed to an estimated 400-500 ppm CO while smoking and consequently have much higher COHb. A necessary consensus, given the variability level of COHb due to environmental CO, suggests an absolute upper limit of normal COHb of 3 % for non-smokers and 10 % for heavy smokers [11].
CAUSES OF RAISED COHb
The amount of COHb in blood is determined principally by the amount of CO in blood. The source of the CO in blood is both endogenous (heme catabolism) and environmental (CO content of inspired air) so that the causes of raised COHb can be addressed under two main headings:
- Increased endogenous production of CO
- Breathing air polluted with high CO content – carbon-monoxide poisoning
Increased endogenous production of CO
Increased endogenous production of CO is a feature of any condition associated with increased heme catabolism. The hemolytic anemias are a group of conditions of variable etiology whose common pathological feature is increased rate of red-cell destruction (hemolysis).
Increased red-cell destruction results in increased heme catabolism and therefore increased production of CO. The severity of hemolysis correlates closely to CO production and measured COHb [12, 13].
In general, COHb increases due to hemolysis are of the order of only 2-3 %, but they can be higher. In one series of 75 neonates suffering hemolytic jaundice [14], five had COHb values in excess of 4 % and one baby with severe hemolysis had a level of 8.3 %. (The normal neonatal COHb range determined for this study was 0.15-0.75 %.)
It is rare for COHb to exceed 10 % in non-smokers, even in the most severe hemolytic episodes.
Slight increase in COHb – so slight to be of little clinical significance per se – is often a feature of severe inflammatory disease, e.g. sepsis, pneumonia [15]. It is thus a relatively common finding in critically ill patients [16].
The mechanism of this increase is thought to be increased expression of heme oxygenase (the enzyme responsible for CO production) induced by inflammatory cytokines [17].
Increased endogenous production of CO can arise independently of heme catabolism. Methylene chloride (dichloromethane) is a toxic organic solvent with wide application, including paint remover, degreaser and aerosol propellant. The toxicity of methylene chloride is due in part to its in vivo metabolism in the liver to CO.
Subjects who inhale toxic amounts of methylene chloride vapor, usually a result of working in poorly ventilated conditions, have raised COHb caused by increased production of CO. COHb levels, which can be severe enough to threaten life, correlates well with methylene chloride exposure levels [18].
Breathing air polluted with high CO content – carbon-monoxide poisoning
This is clinically the most significant cause of increased COHb for two reasons. First it is a more common cause of increased COHb than endogenous production of CO, and secondly it can result in a much more severe increase in COHb.
Most clinical requests for measurement of COHb are made in the context of known or suspected acute or chronic carbon-monoxide poisoning.
Epidemiology of carbon-monoxide poisoning
Deliberate or accidental poisoning by carbon monoxide remains a significant problem. In the US, it accounts for an estimated 40,000 emergency room attendances and between 5,000 and 6,000 deaths each year [19].
Most of these are suicides, usually the result of deliberate exposure to motor-vehicle exhaust, but still 600 deaths a year result from accidental exposure to carbon monoxide from a wide variety of sources. In the UK, CO is responsible for 50 deaths and 200 serious injuries every year [20].
Internationally, CO may be responsible for more than half of all fatal poisonings worldwide [21]. Low-grade chronic CO poisoning is associated with non-specific symptoms and requires a high degree of suspicion for diagnosis, and most authorities believe many cases remain undiagnosed or misdiagnosed [22].
Sources of carbon monoxide
Carbon monoxide is a ubiquitous product of incomplete combustion of hydrocarbons. Common sources of CO in cases of poisoning include house fire, motor-vehicle exhaust and faulty domestic heating systems.
Less commonly, gas ovens, paraffin (kerosene) heaters and even charcoal briquettes, e.g. for use on barbeques, have been implicated.
Clearly a closed or poorly ventilated environment is an important contributory factor in most cases, but it remains possible to suffer severe, even fatal, CO poisoning in the outdoors if close enough to a rich source of CO, e.g. swimming near boat exhaust [23].
Effect of CO exposure on COHb levels
The amount of COHb in blood is a function of both inspired CO concentration (parts per million, ppm) and duration of exposure.
During exposure to a fixed CO concentration, COHb levels increase rapidly over the first 2 hours, then begin to plateau at around 3 hours, reaching an equilibrium steady state at 4-6 hours. Table I describes the relationship between CO exposure and equilibrium COHb.
|
TABLE I: Data relating CO exposure to % COHb and CO concentration in specific environments
CONSEQUENCES OF RAISED COHb
Toxicity of carbon monoxide
The toxicity of CO is due in part to the effect that hemoglobin binding of CO has on the oxygen-carrying capacity of blood. Affinity of hemoglobin for CO is 200-250 times greater than that for oxygen [9, 20, 23, 24].
CO displaces oxygen from hemoglobin and thus COHb effectively reduces the oxygen-carrying capacity in a dose-dependant manner. In addition, binding of CO by Hb at the first of the four heme sites has an effect on its quaternary structure that results in decreased affinity for oxygen at the remaining three sites.
This effect is evident in a shift of the hemoglobin dissociation curve to the left (Figure I) and results in reduced release of oxygen from hemoglobin at the tissues. The combined effect of a reduced oxygen-carrying capacity and reduced release of oxygen to tissue leaves tissues effectively starved of oxygen (hypoxic).
Organs like the brain and heart, whose normal oxygen consumption is by comparison with other organs relatively high, are particularly sensitive to the relative anoxia induced by increased COHb.
Fetal Hb exhibits an even higher affinity for CO than adult Hb, so that since CO diffuses readily across the placental membrane, the developing fetus is particularly vulnerable to tissue anoxia in cases of maternal CO exposure [26].
If increased production of COHb were, as was once supposed, the only mechanism involved in CO toxicity, then the severity of symptoms would be accurately predicted by the level of COHb, but this is not always the case.
It is now clear that "free" CO dissolved in blood plasma enters tissues and competes with oxygen for sites on tissue-cell heme proteins such as myoglobin, peroxidase and the cytochrome enzymes with a variety of pathological effects independent of hemoglobin CO binding [20].
Clinical sign and symptoms of carbon-monoxide poisoning
A high index of suspicion is required to entertain a diagnosis of carbon-monoxide poisoning unless CO exposure is certain, because all symptoms of mild-to-moderate poisoning are non-specific. The classic "cherry-red" skin color of carbon-monoxide poisoning is in fact not usually evident.
The most common symptoms: headache, dizziness and confusion reflect the marked sensitivity of the brain to relative anoxia. Nausea and vomiting are also common.
Affected patients may be breathless, particularly on exertion, and have clinical signs (tachycardia, tachypnea) indicating compensation for the oxygen deficit.
In more severe cases there are frank signs and symptoms of cardiac involvement, including palpitations, hypotension, ischemic chest pain (angina) and even myocardial infarction. Convulsions and coma occur in severe toxicity.
Exposure to carbon monoxide at concentrations greater than 1,900 ppm is immediately fatal.
A raised COHb in the absence of a disease process associated with the hemolytic process is diagnostic of carbon-monoxide poisoning; the actual level correlates with the severity of symptoms in the majority of cases (Table II).
|
||||||||||||
|
TABLE II: Relationship between % CO-Hb and symptoms
SOME ILLUSTRATIVE CASE HISTORIES
Case history 1: Severe CO poisoning with only marginally raised COHb
This case [27] concerns a 13-year-old boy who started his motorbike in the family garage. Before he could get to the garage door he was overcome by the exhaust fumes and collapsed. He was found unconscious around 9 hours after he was last seen, wedged between the family car and the unopened garage door.
Although by now there was no evidence of CO exposure, e.g. running motor or smell of exhaust, the moribund boy was suffering the effects of severe CO inhalation. After initial assessment at the local hospital, his respiration, already "rapid and labored" on admission, deteriorated and he was intubated and transferred to a tertiary referral center, some 13 hours after he was found.
The cause of his continuing unconscious state remained a mystery at this time. On admission to the second hospital, blood was sampled for COHb estimation. The laboratory reported a COHb of 4.9 %.
The boy remained deeply comatose for 10 days and was dependent on mechanical ventilation for 11 days. During this time, convulsions were frequent. Other significant complications included acute renal failure and severe muscle necrosis. Neurological recovery was gradual.
Although apparently alert by day 12, at first he was unable to recognize family members, unable to speak, had no memory and his control of movement was greatly restricted.
At six weeks, his memory had improved sufficiently to recall the events of the day of the accident, and he was able to confirm exposure to motorbike exhaust fumes. Eight weeks after admission he was eventually discharged to a rehabilitation unit, still with some restriction of movement of his lower limbs.
The CO exposure had left him with some impairment of short- and long-term memory, reduced ability to concentrate and a probable IQ deficit.
This is a case history of severe, near-fatal CO exposure with typically severe neurological sequelae. Such severe exposure would normally be associated with very high COHb, possibly in the range of 40-50 %, certainly greater than 20 %. Why then was the COHb only 4.9 %? After all, most smokers endure a COHb > 5 %.
The answer lies in the temporal relationship between exposure and blood sampling and highlights an important limitation of COHb measurement for diagnosis of CO poisoning.
COHb has a half-life of only 4 hours when breathing room air; this is reduced to 90 minutes when breathing 100 % oxygen and less than 30 minutes if hyperbaric oxygen is instituted [10]. This is the rationale for the use of 100 % oxygen or hyperbaric oxygen in the treatment of CO poisoning.
However, it also means that if there is more than a few hours delay between exposure and sampling of blood, COHb will not accurately reflect exposure. In this case, 13 hours elapsed between the time the boy was found and the time blood was sampled.
Given a half-life of 4 hours, this is time enough for COHb to drop from a peak of say 40 % to 5 %. Whilst a raised COHb always indicates CO poisoning, a normal COHb is not sufficient to exclude a diagnosis of CO poisoning if there has been delay between exposure and blood sampling, especially if oxygen therapy has been administered.
Case history 2: An unusual cause of raised COHb
The patient was a critically ill 41-year-old non-smoking male who had been transferred from his local intensive care unit to a tertiary referral center for continued management of large bilateral spontaneous adrenal hemorrhage [28].
On day 6 after referral, blood gas analysis revealed a COHb of 3.9 %, which increased to a maximum of 6.4 % three days later and fluctuated between 1.7 % and 5.6 % for the following two weeks.
Despite repeated transfusion of fresh frozen plasma to correct the presumed causative coagulopathy, internal bleeding continued and on day 14 at exploratory laparotomy, a 4,000 mL hematoma was removed. Biopsy of the adrenal gland revealed a benign tumor (pheochromocytoma) as the cause of bleeding.
Both before referral and for the following 14 days, repeated transfusion of packed red cells were needed to maintain hemodynamic stability. Despite continued intensive care and several further surgical interventions, including adrenalectomy, the patient’s condition deteriorated and he died 58 days after referral.
The principle cause of raised COHb in this case was increased endogenous production of carbon monoxide. This was due to the ongoing degradation of hemoglobin within the retroperitoneal hematoma formed as a result of accumulating blood.
An additional contributory factor may have been the repeated red-cell transfusions. There is evidence that some packed red cells for transfusion may have COHb levels as high as 12 % [29].
Case history 3: COHb does not always correlate well with symptoms [30]
After traveling in a poorly maintained family car for nearly an hour, one of the five passengers, a normally boisterous two-year-old girl fell asleep and was sufficiently unresponsive to raise concern.
She was driven direct to a nearby pediatric emergency room where she was found to be flaccid and responded only to deep painful stimulation with a cry and sluggish opening of her eyes (Glasgow Coma Score 8).
Apart from this reduced level of consciousness, physical examination revealed no abnormalities and a presumptive diagnosis of carbon-monoxide poisoning was made. Within 15 minutes of starting 100 % oxygen therapy the girl was awake. COHb of blood sampled before therapy was 35 %.
After two hours of oxygen therapy, COHb was 7 % and the little girl was fully alert. (GCS 15).
Blood was also sampled for COHb from four other occupants of the car; two children aged two and seven years and two female adults. COHb of the two children was 33.6 % and 34.7 % and the adults had COHb of 18.4 % and 16.1 %. Both children were asymptomatic, one of the adults complained of slight headache and the other of light-headedness.
This case study demonstrates that simultaneous exposure to the same CO source does not necessarily result in the same measured level of COHb, and symptoms manifested by individuals exposed to the same CO source may be dissimilar, despite almost identical COHb results.
SUMMARY
It is difficult to establish a normal range for COHb because the amount of COHb in blood is crucially dependent on variable levels of environmental carbon-monoxide pollution.
Unequivocal increase in COHb indicates either a hemolytic process or more commonly carbon-monoxide poisoning. Increased COHb reduces tissue oxygenation but this is not the only mechanism of CO toxicity. Laboratory measurement of COHb is the only routinely available blood test for diagnosis of CO poisoning.
It provides useful though limited prognostic information in such cases.
References+ View more
- Ranney H M, Sharma V. Structure and Function of Hemoglobin. In Beutler E, Lichtman M et al (eds) Willimas’s Hematology (6th edition) McGraw Hill, 2000: 345-53
- Sjostrand, T. Endogenous formation of carbon monoxide in man under normal and pathological conditions. Scand J Clin Invest. 1949; 1: 201-06
- Coburn RF, Williams WJ, Forster RE. Effect of erythrocyte destruction on carbon monoxide production in man. J Clin Invest 1964; 43: 1098-1103
- Wolff D, Bidlack W. The formation of carbon monoxide during peroxidation of microsomal lipids. Biochem and Biophys Research Communications. 1976; 73: 850-57
- Prabhakar N, Dinerman JL, Agani FH, Snyder SH. Carbon monoxide: a role in carotid body chemoreception. Proc Natl Acad Sci 1995; 92: 1994-97
- Ingi T, Ronnett GV. Direct demonstration of a physiological role for carbon monoxide in olfactory receptor neurons. J Neuroscience 1995; 15: 8214-22
- Kozma F, Johnson RA, Zhang F et al. Contribution of endogenous carbon monoxide to regulation of diameter in resistance vessels. Am J Physiol 1992; 276: R1087-R1094
- Acavedo CH, Ahmed A. Hemeoxgenase-1 inhibits human myometrial contractility via carbon monoxide and is upregulated by progesterone during pregnancy. J Clin Invest 1998; 101: 949-55
- Johnson RA, Kozma F, Colombari E. Carbon monoxide: from toxin to endogenous modulator of cardiovascular functions. Brazilian J Med & Biol Research 1999; 32: 1-14
- Ernst A, Zibrak J. Carbon monoxide poisoning NEJM 1998; 339: 1603-08
- Marshall M, Kales S, Christiani D, Goldman R. Are reference intervals for carboxyhemoglobin appropriate? A survey of Boston area laboratories. Clin Chem 1995; 41: 1434-38
- Coburn RF, Williams WJ, Kahn SB. Endogenous carbon monoxide production in patients with hemolytic anemia. J Clin Invest 1966 45: 460-68
- Engel R, Rodkey F, Krill C. Carboxyhemoglobin levels as an index of hemolysis. Pediatrics 1971; 47: 723
- Necheles T, Rai U, Valaes T. The role of hemolysis in neonatal hyperbilirubinemia as reflected in carboxyhemoglobin values. Acta Paediatr Scand. 1976; 65: 361-67
- Yasuda H, Yamaya M, Ohrui T, Sasaki H. Increased blood carboxyhaemoglobin concentrations in inflammatory pulmonary diseases. Thorax 2002; 57: 779-83
- Morimatsu H, Takahashi T, Maeshima K et al. Increased heme catabolism in critically ill patients: Correlation among exhaled carbon monoxide, arterial carboxyhemoglobin and serum bilirubin IX {alpha} concentrations. Am J Physiol Lung Cell Mol Physiol. (EPub) 2005 Aug 12th doi:/0.1152/ajplung.00031.2005
- Mines D. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol 1997; 37: 517-54
- Shusterman D Quinlan P, Lowengart R, Cone J. Methylene chloride intoxication in a furniture refinisher. J Occupational Medicine 1990; 32: 451-54
- Kao L. Nanagas K. Carbon monoxide poisoning. Emerg Clin N Amer 2004; 22: 985-1018
- Dept. of Health (UK) Carbon monoxide: The forgotten killer. Letter from Chief Medical Officer 2002: PL/CMO/2002/2
- Raub J, Mathieu-Nolf M, Hampson N, Thom S. Carbon monoxide poisoning – a public health perspective. Toxicology 2000; 145: 1-14
- Harper A. Croft-Baker J Carbon monoxide poisoning: undetected by both patients and their doctors. Age and Ageing. 2004; 33: 105-09
- Anon. Houseboat-associated carbon monoxide poisoning on Lake Powell – Arizona and Utah 2000. JAMA 2001; 285: 530-31
- Anon. Carbon Monoxide (Chapter 5.5) In: Air Quality Guidelines for Europe 2nd Ed 2000. WHO Regional Office for Europe Copenhagen Denmark ISBN: 92 890 1358 3
- Winter PM, Miller JN. Carbon monoxide poisoning. JAMA 1976; 236: 1052-1505
- Farrow J, Davis G, Roy T et al. Fetal death due to non-lethal maternal carbon monoxide poisoning. J Forensic Science 1990; 35: 1448-52
- Zimmerman S. Carbon monoxide poisoning. Pediatrics 1981; 68: 215-24
- Ziemann-Gimmel P, Schwartz D. Increased carboxyhemoglobin in a patient with a large retroperitoneal hematoma. Anesth Analg 2004; 99: 1800-02
- Ehlers M, McCloskey D, Devejian N. Alarming levels of carboxyhemoglobin in a unit of banked blood. Anesth Analg 2003; 97: 289-90
- Sanchez R, Fosarelli P, Felt B et al. Carbon monoxide poisoning due to automobile exposure: Disparity between carboxyhemoglobin levels and symptoms of victims. Pediatrics 1988; 82: 663-65
References
- Ranney H M, Sharma V. Structure and Function of Hemoglobin. In Beutler E, Lichtman M et al (eds) Willimas’s Hematology (6th edition) McGraw Hill, 2000: 345-53
- Sjostrand, T. Endogenous formation of carbon monoxide in man under normal and pathological conditions. Scand J Clin Invest. 1949; 1: 201-06
- Coburn RF, Williams WJ, Forster RE. Effect of erythrocyte destruction on carbon monoxide production in man. J Clin Invest 1964; 43: 1098-1103
- Wolff D, Bidlack W. The formation of carbon monoxide during peroxidation of microsomal lipids. Biochem and Biophys Research Communications. 1976; 73: 850-57
- Prabhakar N, Dinerman JL, Agani FH, Snyder SH. Carbon monoxide: a role in carotid body chemoreception. Proc Natl Acad Sci 1995; 92: 1994-97
- Ingi T, Ronnett GV. Direct demonstration of a physiological role for carbon monoxide in olfactory receptor neurons. J Neuroscience 1995; 15: 8214-22
- Kozma F, Johnson RA, Zhang F et al. Contribution of endogenous carbon monoxide to regulation of diameter in resistance vessels. Am J Physiol 1992; 276: R1087-R1094
- Acavedo CH, Ahmed A. Hemeoxgenase-1 inhibits human myometrial contractility via carbon monoxide and is upregulated by progesterone during pregnancy. J Clin Invest 1998; 101: 949-55
- Johnson RA, Kozma F, Colombari E. Carbon monoxide: from toxin to endogenous modulator of cardiovascular functions. Brazilian J Med & Biol Research 1999; 32: 1-14
- Ernst A, Zibrak J. Carbon monoxide poisoning NEJM 1998; 339: 1603-08
- Marshall M, Kales S, Christiani D, Goldman R. Are reference intervals for carboxyhemoglobin appropriate? A survey of Boston area laboratories. Clin Chem 1995; 41: 1434-38
- Coburn RF, Williams WJ, Kahn SB. Endogenous carbon monoxide production in patients with hemolytic anemia. J Clin Invest 1966 45: 460-68
- Engel R, Rodkey F, Krill C. Carboxyhemoglobin levels as an index of hemolysis. Pediatrics 1971; 47: 723
- Necheles T, Rai U, Valaes T. The role of hemolysis in neonatal hyperbilirubinemia as reflected in carboxyhemoglobin values. Acta Paediatr Scand. 1976; 65: 361-67
- Yasuda H, Yamaya M, Ohrui T, Sasaki H. Increased blood carboxyhaemoglobin concentrations in inflammatory pulmonary diseases. Thorax 2002; 57: 779-83
- Morimatsu H, Takahashi T, Maeshima K et al. Increased heme catabolism in critically ill patients: Correlation among exhaled carbon monoxide, arterial carboxyhemoglobin and serum bilirubin IX {alpha} concentrations. Am J Physiol Lung Cell Mol Physiol. (EPub) 2005 Aug 12th doi:/0.1152/ajplung.00031.2005
- Mines D. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol 1997; 37: 517-54
- Shusterman D Quinlan P, Lowengart R, Cone J. Methylene chloride intoxication in a furniture refinisher. J Occupational Medicine 1990; 32: 451-54
- Kao L. Nanagas K. Carbon monoxide poisoning. Emerg Clin N Amer 2004; 22: 985-1018
- Dept. of Health (UK) Carbon monoxide: The forgotten killer. Letter from Chief Medical Officer 2002: PL/CMO/2002/2
- Raub J, Mathieu-Nolf M, Hampson N, Thom S. Carbon monoxide poisoning – a public health perspective. Toxicology 2000; 145: 1-14
- Harper A. Croft-Baker J Carbon monoxide poisoning: undetected by both patients and their doctors. Age and Ageing. 2004; 33: 105-09
- Anon. Houseboat-associated carbon monoxide poisoning on Lake Powell – Arizona and Utah 2000. JAMA 2001; 285: 530-31
- Anon. Carbon Monoxide (Chapter 5.5) In: Air Quality Guidelines for Europe 2nd Ed 2000. WHO Regional Office for Europe Copenhagen Denmark ISBN: 92 890 1358 3
- Winter PM, Miller JN. Carbon monoxide poisoning. JAMA 1976; 236: 1052-1505
- Farrow J, Davis G, Roy T et al. Fetal death due to non-lethal maternal carbon monoxide poisoning. J Forensic Science 1990; 35: 1448-52
- Zimmerman S. Carbon monoxide poisoning. Pediatrics 1981; 68: 215-24
- Ziemann-Gimmel P, Schwartz D. Increased carboxyhemoglobin in a patient with a large retroperitoneal hematoma. Anesth Analg 2004; 99: 1800-02
- Ehlers M, McCloskey D, Devejian N. Alarming levels of carboxyhemoglobin in a unit of banked blood. Anesth Analg 2003; 97: 289-90
- Sanchez R, Fosarelli P, Felt B et al. Carbon monoxide poisoning due to automobile exposure: Disparity between carboxyhemoglobin levels and symptoms of victims. Pediatrics 1988; 82: 663-65
May contain information that is not supported by performance and intended use claims of Radiometer's products. See also Legal info.


Acute care testing handbook
Get the acute care testing handbook
Your practical guide to critical parameters in acute care testing.
Download now
Related webinar
Evolution of blood gas testing Part 1
Presented by Ellis Jacobs, PhD, Assoc. Professor of Pathology, NYU School of Medicine.
Watch the webinar