Printed from acutecaretesting.org
September 2013
Developing quality control strategies based on risk management: The CLSI EP23 guideline




INTRODUCTION
The modern clinical laboratory is a complex environment with significant potential for error. Technologists perform hundreds of different tests using a variety of sophisticated analytical instrumentation.
These are the questions that challenge a laboratory director. Risk management principles are a way to map our testing, define our weaknesses, and identify the right control processes that can detect and prevent errors in our laboratory.
RISK
Risk management is the systematic application of management policies, procedures and practices to the tasks of analyzing, evaluating, controlling and monitoring risk [1]. Risk is essentially the potential for an error to occur.
There is thus a spectrum of risk from high level to lower level. One can never achieve zero risk, since there will always be the potential for error. But, through detection and prevention, we can reduce the chance of risk to a clinically acceptable level.
Laboratories conduct a number of activities every day that could be considered risk management. Technologists evaluate the performance of instrumentation before implementing it for patient care.
Controls are analyzed each day and when controls fail, technologists troubleshoot tests and take action to prevent errors from recurring. The laboratory director responds to physici
an complaints and estimates harm to a patient if incorrect results are reported.So, risk management is not a new concept, just a formal term for what we are already doing each day in the laboratory.
There is no perfect device; otherwise we will all be using it! No instrument is fool-proof. Any test can and will fail under the right conditions. Any discussion of risk must start with what can go wrong with a test.
Laboratory errors can arise from many sources including; the environment, the specimen, the operator and the analysis. The environment can affect reagent and instrumentation.
Temperature; heat and cold, as well as light can compromise reagents. Specimens may be improperly transported or handled prior to analysis. Bubbles, clots and drugs may interfere with a test.
Operators may fail to follow procedures, incorrectly time a reaction, inaccurately pipette a sample, or make other testing mistakes. Instruments may fail during analysis or error may occur in reporting a result across an electronic interface.
There are thus a number of ways that errors can arise in the laboratory.
CONTROL PROCESSES
Controls have historically been used to detect and prevent errors. The concept of utilizing controls in the clinical laboratory rose from the 1950s’ industrial model of quality.
The use of liquid quality control has advantages and disadvantages. The control is a stabilized sample of known concentration that, when analyzed periodically over time, can assess the ongoing performance of a test system.
Since the control is analyzed like a patient sample, performance of the control is assumed to mimic performance of patient samples on the analyzer.
However, the process of stabilizing a control sample can change the matrix, so controls do not always reflect test performance of patient samples. Controls do a great job at detecting systematic errors; errors that affect every test in a constant and predictable manner.
Systematic errors include reagent deterioration, incorrect operator technique (e.g., dilution or pipette settings), and wrong calibration factors. Controls, however, do a poor job at detecting random errors; errors which affect individual samples in a random and unpredictable fashion, like clots, bubbles or interfering substances.
For batch analysis, controls are analyzed before and after patient testing. If the control performance is acceptable, then patient results are released. But, with an increased pressure for faster turnaround times, patient results may be released continuously as soon as the result is available from our automated instruments.
When a control fails to achieve expected performance, staff must determine at what point an analyzer failed, troubleshoot, and fix the instrument. Patient specimens may need to be reanalyzed and corrected reports sent to physicians if the results are significantly affected.
When a laboratory corrects a result, physicians lose faith in the reliability of the laboratory. We need to get to fully automated analyzers that allow for continuous release of results, eliminate errors upfront, and provide assured quality with every sample.
Until that time, we need a robust control plan to ensure result quality.
Newer instrumentation offers a variety of control processes in addition to the analysis of liquid control samples. There is “on-board” or instrument quality control that can be analyzed at timed intervals automatically.
Manufacturers have also engineered processes that are built-in or system checks to automatically detect and prevent certain errors from occurring. Clot and bubble detection in blood gas analyzers are examples of this type of manufacturer control process.
No single quality control procedure can cover all devices, since devices may differ in design, technology, function and intended use [3]. Quality control practices developed over the years (i.e., two levels of controls each day of testing) have provided laboratories with some degree of assurance that results are valid.
Newer devices have built-in electronic controls and “on-board” chemical and biologic control processes. Quality control information from the manufacturer increases the operator’s understanding of device overall quality assurance requirements, so that informed decisions can be made regarding suitable control procedures [3].
Laboratory directors have the ultimate responsibility for determining the appropriate quality control procedures for their laboratories. Manufacturers of in vitro devices have the responsibility for providing adequate information about the performance of devices, means to control risks, and verify performance within specification.
In practice, quality control is a shared responsibility between manufacturers and users of diagnostic devices [3].
The Clinical and Laboratory Standards Institute (CLSI) EP23-A document [4]: laboratory quality control based on risk management provides guidance for laboratories to determine the optimum balance between manufacturer built-in, engineered control processes and traditional analysis of liquid controls.
The EP23 guideline describes good laboratory practice for developing a quality control plan based on manufacturer’s information, applicable regulatory and accreditation requirements, and the individual healthcare and laboratory setting (Fig. 1).
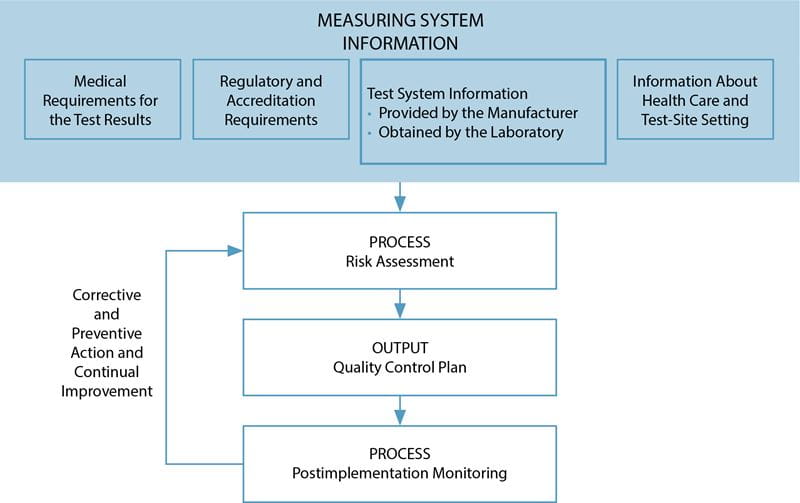
FIG. 1: Process to develop and continuously improve a quality control plan. (Reproduced with permission from the Clinical and Laboratory Standards Institute, Wayne, PA. EP23-A guideline: Laboratory Quality Control Based on Risk Management, 2011, Wayne, PA)
Information about the test system, the medical requirements for the test, local regulatory requirements, and laboratory setting are processed through a risk assessment to develop a quality control plan specific to the laboratory, the device and the patient population.
Once implemented, this plan is monitored for trends and errors, then modified as needed to continuously improve the plan and maintain risk to a clinically acceptable level.
Use of risk management principles allows a laboratory to map their testing, define weaknesses in their procedures, identify appropriate control processes and develop a plan to minimize errors.
AN EXAMPLE OF RISK MANAGEMENT IN PRACTICE
A simple point-of-care test, a serum pregnancy test, can demonstrate risk management principles and how EP23 can help laboratories develop a quality control plan.
For this example, a physician office laboratory wants to implement a rapid pregnancy test to better manage patients while they are in the office. The volume of testing is low, only one or two tests a day.
So, the need for two levels of daily liquid controls will add cost and delay the analysis of patient samples if the office has to wait for control results before patient tests each day.
The manufacturer claims to have incorporated controls within each test, reducing the need for daily liquid controls, that would improve the cost, test and labor efficiency for the physician office. But, the office needs a comparable quality of result.
So, developing a control plan to define the right balance of internal and external controls is needed.
The first step to develop a quality control plan is to gather information about the test. The test device is a two-site, immunochromatography or immunometric assay (Fig. 2).
Sample is added to the test well with a pipette. If the sample is positive for human chorionic gonadotropin (hCG), the protein reacts with colored conjugate-bound antibody (mouse anti-beta subunit hCG antibody-bound conjugate).
The bound conjugate wicks down a paper membrane with the sample and reacts with antibody at the test line (anti-alpha subunit hCG) to form a colored line. Conjugate that does not bind at the test line continues to wick down the membrane and reacts with goat anti-mouse antibodies at the control line.
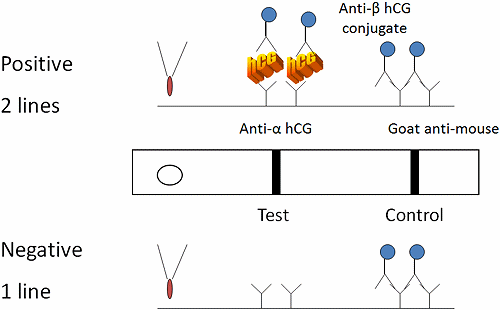
FIG. 2: Two-site immunometric serum hCG assay. Specimen is applied to the test well and wicks through capillary action down the chromatography paper. hCG in the patient’s sample acts to cross-link colored conjugate-bound anti-beta subunit antibody to anti-alpha subunit antibody at the test area. Conjugate that is not bound at the test line reacts with goat anti-mouse antibody at the control area of the test. Development of two lines is interpreted as a positive result, while one line is negative. If no lines are visualized, the control is invalid and the reactivity of the test is in question. Invalid results should repeat the test.
The test is interpreted by the number of colored lines. Two lines are positive (one at the test and one at the control region) while one line is negative (the control region).
If no lines develop, the internal control did not react and the test is invalid. The internal control line verifies sample and reagent wicking on the membrane, adequate sample volume, reagent viability and correct procedure.
A negative procedural control is the background clearing between the test and control areas and verifies adequate wicking and test procedure.
The next step to develop a quality control plan is to create a process map in order to identify weak steps, hazards or risk of errors, in the testing procedure.
A test must be ordered, sample collected, transported to the test kit, and the test must be properly interpreted and the result reported to the clinician.
Within this testing process, errors could occur in the timing of sample collection, collection with the wrong tube additive, interferences from specimen clotting and hemolysis, delays in testing, reagent degradation during shipping or storage, wrong test procedure, timing or interpretation.
For each hazard or potential error identified, the laboratory must define a control process to maintain that risk to an acceptable level. The test must be collected at the right time, too early in pregnancy could give a false negative.
Patients with high suspicion of pregnancy will need to be retested in 48 hours. A false positive result can occur from certain neoplasms that secrete hCG as well as patients with human anti-mouse antibodies (HAMA).
The laboratory will need to educate staff on the limitations of the test. The test requires serum, and use of plasma would have unknown performance on the test. Since hCG is a protein, delays in sample testing and temperature exposure could compromise the test.
In an office setting, this would be of minimal concern if samples are analyzed immediately after collection. So, no additional control processes may be required for sample delays or temperature exposure risks, since the sample is not being transported outside the office.
In addition, sample clots and hemolysis could impede performance, so the laboratory should process the samples promptly, monitor for specimen quality and maintain the centrifuges.
Reagent exposure to extreme temperatures presents a concern over degradation during shipping and ongoing stability during storage. Liquid controls will need to be analyzed to verify reactivity upon receipt of new shipments and storage conditions will need to be monitored in the laboratory.
Periodic analysis of controls can verify stability during storage. Unless daily controls are mandated by the manufacturer, the laboratory could start with daily controls, then back off to weekly and even biweekly or monthly frequency once the laboratory gains more experience with the test and confidence in reagent stability.
Wrong sample volume could be an issue, but the test comes with a calibrated disposable pipette. Use of this pipette minimizes risk of incorrect volume.
The internal controls will detect a wrong procedure, provided that the laboratory uses a timer for appropriate test development.
TABLE I: Quality control plan for a generic serum pregnancy test. The quality control plan is a summary of actions for the laboratory to minimize the risk of errors to a clinically acceptable level when utilizing this test.
– Each new shipment* – Start of a new lot* – Monthly (based on prior experience with test) – Whenever uncertainty exists about test*
– Use proper tube type. – Process specimens promptly (centrifuge maintenance). – Check expiration dates before use. – Proper test interpretation and double-check result reporting. – Monitor storage temperature. (* Manufacturer recommendations) |
The control plan is summarized in Table I. Control plans must be checked to ensure that the plan meets minimum manufacturer recommendations for control performance and complies with local quality regulations.
For this example, the laboratory will analyze liquid controls with each new shipment, at the start of a new lot, monthly (after some experience with the test), and whenever there is uncertainty about the reactivity of the test.
These are recommended in the manufacturer’s package insert. In addition, the laboratory will need to monitor internal controls with each test. Physicians need education on the limitations of the test.
The office should use a timer for test development and use a dedicated transfer pipette for sample application.
A checklist for operator training will need to incorporate these items in addition to checking appropriate tube type (serum collection tube), proper processing and centrifuge maintenance, checking expiration dates before use, monitoring reagent storage conditions, and correct test interpretation.
After implementation, the effectiveness of the quality control plan should be benchmarked and monitored for trends. The laboratory could monitor the rates of internal test control failure, specimen quality concerns or even requests for follow-up or retesting of patients.
SUMMARY
Risk management provides a means of mapping a laboratory’s testing process, identifying weak steps in the process, and optimizing controls to detect and prevent error.References+ View more
- ISO. Medical devices – Application of risk management to medical devices. International Organization for Standardization Publication 14971. Geneva, Switzerland: ISO, 2007; 1-82.
- ISO/IEC. Safety aspects – Guidelines for their inclusion in standards. International Organization for Standardization/International Electrotechnical Commission Guide 51. Geneva, Switzerland: ISO/IEC, 1999: 1-10.
- ISO. Clinical laboratory medicine – In vitro diagnostic medical devices – Validation of user quality control procedures by the manufacturer. International Organization for Standardization Publication 15198. Geneva, Switzerland: ISO, 2004; 1-10.
- Nichols JH, Altaie SS, Cooper G, et al. Laboratory quality control based on risk management. CLSI Publication EP23-A. Wayne, PA.: CLSI, 2011; 31, 18: 1-72.
References
- ISO. Medical devices – Application of risk management to medical devices. International Organization for Standardization Publication 14971. Geneva, Switzerland: ISO, 2007; 1-82.
- ISO/IEC. Safety aspects – Guidelines for their inclusion in standards. International Organization for Standardization/International Electrotechnical Commission Guide 51. Geneva, Switzerland: ISO/IEC, 1999: 1-10.
- ISO. Clinical laboratory medicine – In vitro diagnostic medical devices – Validation of user quality control procedures by the manufacturer. International Organization for Standardization Publication 15198. Geneva, Switzerland: ISO, 2004; 1-10.
- Nichols JH, Altaie SS, Cooper G, et al. Laboratory quality control based on risk management. CLSI Publication EP23-A. Wayne, PA.: CLSI, 2011; 31, 18: 1-72.
May contain information that is not supported by performance and intended use claims of Radiometer's products. See also Legal info.


Acute care testing handbook
Get the acute care testing handbook
Your practical guide to critical parameters in acute care testing.
Download now
Scientific webinars
Check out the list of webinars
Radiometer and acutecaretesting.org present free educational webinars on topics surrounding acute care testing presented by international experts.
Go to webinars