Printed from acutecaretesting.org
October 2007
Lactate and lactic acidosis




Normal lactate production and metabolism [1]
Lactate, the anion that results from dissociation of lactic acid, is a product of glucose metabolism; specifically it is the end product of anaerobic glycolysis. The glycolytic pathway (Fig. 1), which can proceed anaerobically in the cytoplasm of all cells, is a sequence of 13 enzymic reactions in which glucose is converted to pyruvate.
During this conversion, energy-rich adenosine triphosphate (ATP) is generated from adenosine diphosphate (ADP) and reduced nicotinamide adenine dinucleotide (NADH) is generated from oxidized NAD (NAD+).
The final step of anaerobic glycolysis is conversion of pyruvate to lactate by the enzyme lactate dehydrogenase. This last reaction provides a source of NAD+ essential for anaerobic glycolysis to proceed. Production of lactate is the only means for glucose utilization and ATP production in erythrocytes (which have no mitochondrion) and in exercising muscle cells (which have an oxygen debt).
In well-oxygenated tissue cells that contain mitochondrion, pyruvate is not preferentially converted to lactate but rather metabolized to carbon dioxide and water in mitochondria via two integrated metabolic pathways: the citric acid cycle and oxidative phosphorylation.
Conversion of a molecule of glucose to lactate (anaerobic glycolysis) yields just two molecules of ATP, whereas conversion to carbon dioxide and water (aerobic glycolysis) has a much higher energy yield of 38 ATP molecules.
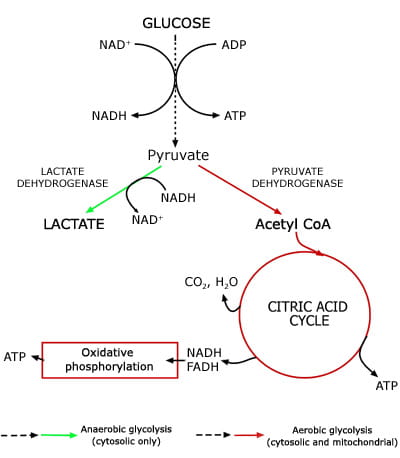
FIG 1: Lactate - a product of anaerobic glycolysis
Although lactate can be produced in all tissues, skeletal muscle, erythrocytes, brain and renal medulla tissues are the principal production sites in health. Normal daily lactate production is of the order of 1500 mmol [1]. There are two main routes of lactate disposal: conversion to pyruvate or elimination in urine (Fig. 2).
Although lactate is freely filtered at the glomerulus, it is almost all reabsorbed and normally <2 % of lactate is removed from the body in urine [3]. The principal route of disposal begins with cytosolic conversion to pyruvate by the enzyme lactate dehydrogenase.
This pyruvate has two principal fates. The first is oxidation to acetyl CoA by the enzyme pyruvate dehydrogenase for ultimate metabolism to carbon dioxide and water via the citric acid cycle and oxidative phosphorylation. The second is conversion to glucose, a process called gluconeogenesis. Oxidation of pyruvate via the citric acid cycle can potentially occur in all cells with mitochondrion, but gluconeogenesis is confined to liver and kidney cortex cells.
For this reason liver and kidneys are the most significant organs for lactate elimination, the liver accounting for disposal of around 60 % of circulating lactate [4] and the kidneys for disposal of 25-30 % [5]. Overall, the body has immense capacity for lactate disposal, which can, if necessary (e.g. following extreme exercise), rise as high as 500 mmol/hour, considerably higher than the basal rate of production.
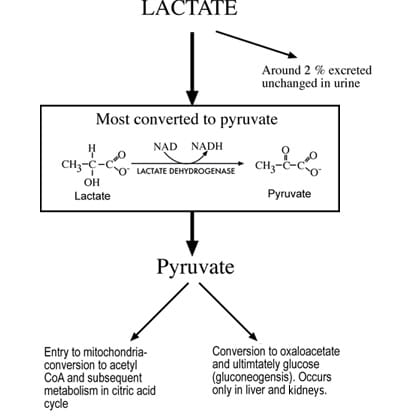
FIG 2: Principal mechanisms of lactate disposal
Normal blood lactate concentration
Lactate produced within erythrocytes cannot be metabolized further and is released to the circulation. In some tissues (e.g. skeletal muscle) lactate may be produced at a faster rate than it can be metabolized and in these circumstances lactate would also be released to circulation.
In health, blood lactate concentration is maintained within the approximate range of 0.5-1.5 mmol/L [6]. This reflects a balance between the rate of lactate release to blood from erythrocytes and other tissue cells and rate of lactate clearance from blood, principally by the liver and kidney.
Exercise represents a physiological process in which this balance is temporarily upset due to the rapid increase in lactate production by muscle cells in temporary oxygen debt. In severe exercise, blood lactate may rise to levels in excess of 20 mmol/L but due to the capacity for rapid lactate disposal, in health this rise is only transitory.
Hyperlactatemia and lactic acidosis
Hyperlactatemia is a pathological state in which resting blood lactate concentration is abnormally high (>1.5 mmol/L). Moderate to severe hyperlactatemia (>3.0 to >5.0 mmol/L) is associated with abnormal accumulation of hydrogen ions (H+) and a resulting tendency to acidosis.
These hydrogen ions are a product of ATP hydrolysis to ADP [2]. In the presence of oxygen, hydrogen ions produced during ATP hydrolysis are utilized in the mitochondrial process of oxidative phosphorylation, but this is often not possible in the context of anaerobic glycolysis associated with hyperlactate production.
Instead, hydrogen ions accumulate in blood, eventually overwhelming the bicarbonate and other buffering systems that maintain blood pH within normal limits (7.35-7.45). The combination of hyperlactatemia and acidosis is called lactic acidosis and although there is no universal agreement for definition of lactic acidosis, the most widely used is blood lactate >5.0 mmol/L in combination with pH <7.35 [7].
Lactic acidosis is the most common cause of metabolic acidosis [8].
Cause of hyperlactatemia and lactic acidosis – general considerations
Hyperlactatemia develops when the rate at which lactate is released from peripheral tissue cells to circulation exceeds the rate at which it is removed from circulation by liver and kidneys. Both increased lactate production and decreased lactate removal/metabolism can be causative.
From a biochemical viewpoint the central problem is usually decreased utilization of pyruvate in oxidative or gluconeogenic pathways. Under these circumstances pyruvate can only be converted to lactate. For example, since oxygen is essential for pyruvate oxidation, any condition that deprives tissues of oxygen can lead to increased production of lactate, which then accumulates in blood at a faster rate than it can be removed by liver and kidneys.
The problem is compounded by acidosis because the capacity of the liver to remove lactate from the circulation is pH dependent and severely impaired by reduced blood pH. In fact, experimental evidence suggests that at blood pH of 7.0 or less, lactate uptake is so impaired that the liver produces more lactate than it consumes [9].
There is some renal compensation because acidosis enhances kidney uptake of lactate [5]. However, this can only compensate for around 50 % of the hepatic loss and acidosis, whatever its cause, can be a major contributory factor in the pathogenesis of hyperlactatemia.
Specific causes of lactic acidosis
Traditionally, lactic acidosis has been divided into two broad etiological categories; Type A and Type B. Type A is lactic acidosis resulting from tissue hypoxia (biochemical mechanism outlined above) and Type B is lactic acidosis occurring in the context of normal tissue perfusion and adequate global tissue oxygenation.
Type A lactic acidosis
Tissue hypoxia and Type A lactic acidosis arise as a result of inadequate perfusion of tissues in hemorrhagic, cardiogenic and septic shock. These conditions associated with volume depletion and/or severe hypotension may be the result of major trauma/surgery or acute medical conditions (e.g. myocardial infarction, systemic infection) causing cardiovascular collapse.
Type A lactic acidosis then is a feature of acute, life-threatening (critical) illness. The conventionally held view that lactic acidosis occurring in the context of sepsis and septic shock is the sole result of tissue hypoxia is now challenged (see below) and for critically ill patients, whose condition is caused or complicated by infection, the distinction between Type A and Type B lactic acidosis is blurred and inappropriate.
Perfusion is not the sole determinant of tissue oxygenation, which also depends on an adequate amount of oxygen in blood. Tissue hypoxia and consequent Type A lactic acidosis can thus occur despite adequate perfusion if the oxygen content of blood or the oxygen-carrying capacity of blood is sufficiently reduced.
This is the mechanism of the Type A lactic acidosis that can occur in patients with severe anemia [10], severe hypoxemia (e.g. respiratory failure) [11] and carbon monoxide poisoning [12]. In practice, anemia and hypoxemia are rarely the sole causes of Type A lactic acidosis. More commonly they are contributory factors in the development of Type A lactic acidosis among patients already predisposed because of inadequate perfusion.
The massive increase in muscular activity that occurs during seizures can cause Type A lactic acidosis due to local muscle tissue hypoxia consequent on a temporary mismatch between oxygen demand and oxygen supply (very similar to the physiological lactic acidosis that occurs during exercise). In common with exercise-induced lactic acidosis, seizure-induced lactic acidosis is self-limiting, spontaneously resolving within a few hours of seizure cessation [13].
Type B lactic acidosis
If lactic acidosis occurs in the context of apparently adequate tissue oxygenation and normal hemodynamics (i.e. normal blood pressure, no volume depletion, normal blood oxygen and oxygen-carrying capacity), it is traditionally termed Type B lactic acidosis.
The vital role of the liver and kidneys for lactate uptake from circulation and subsequent metabolism via the citric acid cycle and gluconeogenesis determines that hepatic and renal disease, whatever its cause, predisposes to mild hyperlactatemia and rarely Type B lactic acidosis. Malignant disease may be associated with lactic acidosis, most cases of Type B lactic acidosis occurring in hematological malignancy (leukemia, lymphoma) [14].
Type B lactic acidosis is a feature of several individually very rare inherited disorders that are characterized by deficiency of specific enzymes involved in lactate metabolism (either gluconeogenesis or pyruvate oxidation). These include pyruvate carboxylase deficiency [15], glucose-6-phosphate dehydrogenase (G6PD) deficiency, fructose-1,6-diphosphatase deficiency [16] and pyruvate dehydrogenase deficiency, the most common [17]. These conditions are collectively referred to as congenital lactic acidosis.
A long list of drugs and toxins can cause lactic acidosis (Table I), and taken together these represent by far the most common cause of Type B lactic acidosis. Biguanides are a class of blood glucose-lowering drugs used in the treatment of diabetes; metformin, the most widely prescribed, has been linked to lactic acidosis [18]. However, in most cases of metformin-associated lactic acidosis, there is some evidence of liver or renal impairment that predisposes to hyperlactatemia.
|
Biguanides (e.g. metformin) |
TABLE I: Some drugs and toxins that can cause lactic acidosis.
Metabolism of ethanol is associated with increased NADH/NAD+ ratio favoring conversion of pyruvate to lactate [2]. Gluconeogenesis is also inhibited so that the combination of moderate hyperlactatemia and hypoglycemia is a not infrequent finding in patients suffering acute effects of alcohol abuse.
Pre-existing alcoholic liver disease exacerbates the acute hyperlactatemia-precipitating Type B lactic acidosis. Salicylate overdose can be associated with lactic acidosis due to the inhibitory effect that salicylate has on oxidative phosphorylation.
A similar mitochondrial effect accounts for the lactic acidosis that occurs in cyanide poisoning. Most HIV patients prescribed anti-retroviral drugs develop mild chronic hyperlactatemia (serum lactate 1.5-3.5 mmol/L) [19], and in a small unpredictable minority this evolves to severe lactic acidosis [20].
Lactic acidosis and sepsis
The notion that lactic acidosis occurring in the context of critical illness is Type A and therefore always indicates tissue hypoxia is clearly over-simplistic. Critically ill patients are as likely as any other to be suffering Type B lactic acidosis and due consideration must be given to the causes of Type B lactic acidosis outlined above when assessing any patient (including those who are critically ill) presenting with raised blood lactate.
Sepsis is an additional and far more significant complicating issue that warrants special attention, not least because it is such a common feature of critical illness. Sepsis is a condition in which the distinction between Type A and Type B lactic acidosis is inappropriate because in some patients with sepsis, lactate accumulates despite adequate tissue oxygenation [21].
A number of mechanisms have been proposed for lactate accumulation in the absence of tissue hypoxia among patients with sepsis. These include reduced clearance of lactate from circulation by liver and kidneys [22]; a specific defect induced by sepsis in the enzyme pyruvate dehydrogenase that impairs pyruvate utilization in the citric acid cycle [23]; and increased pyruvate production [24].
Sepsis and septic shock are associated with a stress response that includes increased release of epinephrine (adrenalin). This hormone stimulates the membrane-bound enzyme Na+/K+-ATPase, which utilizes ATP generated by aerobic glycolysis to provide the energy necessary to "pump" ions into and out of cells. Na+/K+-ATPase stimulation increases aerobic glycolysis and thereby lactate production.
It has been hypothesized that the hyperlactatemia associated with sepsis may be at least in part the result of this epinephrine-stimulated aerobic glycolysis [21].
Whatever the mechanism it is clear that in many whose critical illness is either caused by or complicated by infection, lactic acidosis is not necessarily entirely the result of tissue hypoxia. In these circumstances measures taken to enhance tissue perfusion would be less effective in reducing blood lactate and resolving acidosis.
D-lactic acidosis
This is a rare and very specific form of lactic acidosis confined to those with short-bowel syndrome and those who have undergone jejunoileal bypass surgery [25], a treatment for severe obesity. Lactic acid exists naturally in two isomeric forms: L-lactic acid and D-lactic acid.
In humans lactate is derived from the L-form, but some bacterial species can produce the D-form. In the aforementioned patient group, carbohydrate malabsorption can lead to abnormal increase in the gut of D-lactate-producing bacterial species. The D-lactic acid is released to the gut, absorbed and accumulates in blood.
Susceptible patients suffer episodes of D-lactic acidosis usually following ingestion of a high-carbohydrate meal. Since routine methods of lactate measurement detect only the L-isomer, blood lactate is falsely normal and the only biochemical clue to the condition is unexplained high anion gap acidosis.
Clinical significance of hyperlactatemia
Clinical signs
Mild to moderate hyperlactatemia (blood lactate <5 mmol/L) causes no specific signs and symptoms. As lactate levels rise above 5 mmol/L, there is an increasing risk of the clinical manifestations of lactic acidosis, which are tachycardia (increased heart rate) tachypnea (increased respiratory rate) and alteration in mental status that can range from mild confusion to coma [1]. The deep sighing respiration (Kussmaul’s respiration), a compensatory response to any form of metabolic acidosis, may be evident.
Prognostic value of lactate measurement in critical care
The traditional use of lactate measurement as a marker of inadequate tissue perfusion and tissue hypoxia in critical care is somewhat limited by the increasing realization over the past decade that trauma victims and others suffering potential critical illness may have raised lactate despite adequate tissue perfusion.
Notwithstanding this limitation, lactate measurement has value in the emergency room and intensive care unit because, irrespective of the mechanism of hyperlactatemia, blood lactate concentration predicts morbidity and mortality.
The prognostic value of lactate measurement has been confirmed in many patient groups.
For example, in a prospective study of 76 major trauma victims serial lactate measurements were made over the first 48 hours after admission [26]. All 27 patients whose lactate normalized within 24 hours survived but only three of 22 patients whose lactate remained raised at 48 hours survived.
In a study of 1278 patients with infection, mortality was 4.9 % among those whose lactate was less than 2.5 mmol/L on admission compared with 36 % among those whose lactate was >4mmol/L [27]. Finally, an early study of 233 critically ill patients suffering various forms of shock revealed a mortality rate of 67 % among those whose lactate was >3.82 mmol/L compared with only 25 % for those whose lactate was <3.82 mmol/L [28].
These and many other studies have demonstrated that raised lactate in the context of trauma, sepsis and critical illness generally is a poor prognostic sign indicating the need for immediate and intensive resuscitative measures. If these are effective in reducing blood lactate within 24-48 hours, the chances of survival can be greatly increased [29].
A recent expert committee concluded that there is good evidence that point-of-care lactate testing leads to improved outcomes for patients in a variety of critical care settings [30].
Sampling requirements for lactate measurement
Although some authorities recommend arterial blood for lactate measurement, venous blood is a suitable alternative; results are not clinically significantly different [31]. Use of a tourniquet during blood collection should be avoided because it can cause falsely increased lactate. Heparin is a suitable anticoagulant.
In vitro glycolysis and therefore lactate production continues after sampling so that lactate concentration increases by 30 % in just 30 minutes if kept at room temperature. For this reason blood must be analyzed immediately it is sampled. If there is to be any delay, the sample must be stored on ice to inhibit ongoing glycolysis. Preanalytical errors associated with lactate measurement are the subject of a recent review [32].
Summary
Lactate is a normal product of anaerobic glycolysis. The principal clinical significance of lactate is that in severe acute illness anaerobic glycolysis is often increased – a result of tissue hypoxia. As a consequence blood lactate concentration increases.
The clinical value of lactate as an indicator of tissue hypoxia is diminished by the fact that hyperlactatemia can occur despite normal tissue perfusion and adequate oxygen delivery if, for example, mechanisms of lactate metabolism/disposal are compromised. Irrespective of the cause, hyperlactatemia is often associated with reduction in blood pH and the resulting condition lactic acidosis.
Despite the lack of specificity for tissue hypoxia, lactate is a valuable prognostic marker in all forms of critical illness and a useful diagnostic tool in the investigation of patients presenting with unexplained metabolic acidosis.
References+ View more
- Mordes J P, Rossini AA. Lactic acidosis. In: Irwin and Rippe’s Intensive Care Medicine (4th edition) Ed: Irwin R, Cera FB, Rippe JM. Philadelphia: Lippincott-Raven, 1999.
- Fall P, Szerlip H. Lactic acidosis: from sour milk to septic shock. J Intensive Care Med 2005; 20: 255-71.
- Yudkin J, Cohen RD. The contribution of the kidney to the removal of lactic acid load under normal and acidotic conditions in the conscious rat. Clin Sci Mol Med 1975; 48: 121-31.
- Cohen RD, Woods HF. Clinical and biochemical aspects of lactic acidosis. Oxford: Blackwell Science.
- Bellomo R. Bench to bedside review: lactate and the kidney. Critical Care 2002; 6: 322-26.
- Sacks DB. Carbohydrates. In: Tietz Textbook of Clinical Chemistry and Molecular Diagnostics (4th edition). St. Louis: Elsevier, 2006.
- Mizcock B. Controversies in lactic acidosis: implications in critically ill patients. JAMA 1987; 258: 497-501.
- Cassaletto J. Differential diagnosis of metabolic acidosis. Emerg Med Clin N Amer. 2005; 23: 771-87.
- Lloyd MH, Iles RA et al. The effect of simulated metabolic acidosis on intracellular pH and lactate metabolism in the isolated perfused rat liver. Clin Sci Mol Med 1973; 45: 543-49.
- Essex DW, Jun DK, Bradley TP. Lactic acidosis secondary to severe anemia in a patient with paroxysmal nocturnal hemoglobinuria. Am J Hematol 1998; 55:110-11.
- Aberman A, Hew E. Lactic acidosis presenting as acute respiratory failure. Am Rev Respir Dis 1978 118: 961-63.
- Foster M, Goodwin S et al. Recurrent life-threatening events and lactic acidosis caused by chronic carbon monoxide poisoning in an infant. Pediatrics 1999; 104: e34-e35.
- Orringer CE, Eustace JC et al. Natural history of lactic acidosis after grand-mal seizures: a model for the study of an anion gap acidosis not associated with hyperkalemia. New Eng J Med 1977; 297: 796-99.
- Freidenburg AS, Brandoff DE et al. Type B lactic acidosis as a severe metabolic complication in lymphoma and leukemia: a case series from a single institution and literature review. Medicine 2007; 86: 225-32.
- Farrell DF Clark AF et al. Absence of pyruvate decarboxylase in man: A cause of congenital lactic acidosis. Science 1975; 187: 1082-84.
- Rallison ML, Meikle AW et al. Hypoglycemia and lactic acidosis associated with fructose-1,6 diphosphatase deficiency. J Pediatrics 1979; 94: 933-36.
- Blanco-Barca O, Gomez-Lado C et al. Pyruvate dehydrogenase deficit associated to the C515T mutation in exon 6 of the E1alpha gene. Rev Neurol 2006; 43: 341-45.
- Stacpoole PW. Metformin and lactic acidosis - guilt by association? Diabetes Care 1998; 21:1587-88.
- John M, Moore CB et al. Chronic hyperlactatemia in HIV-infected patients taking antiretroviral therapy. AIDS 2001; 15: 717-23.
- Bonarek M, Bonnet F et al. Severe lactic acidosis in HIV infected patients treated by nucleoside analog reverse-transcriptase inhibitors – a report of 9 cases. Rev Med Interne 2003; 24: 11-16.
- James JH, Luchette FA et al. Lactate is an unreliable indicator of tissue hypoxia in injury or sepsis. Lancet 1999; 354: 505-08.
- Levraut J, Ciebiera J-P et al. Mild hyperlactatemia in stable septic patients is due to impaired lactate clearance rather than over production. Am J Respir Crit Care Med 1998; 157: 1021-26.
- Vary TC, Siegel JH et al. Effect of sepsis on activity of pyruvate dehydrogenase complex in skeletal muscle and liver. Am J Physiol 1986; 250: E634-E640.
- Gore D, Jahoor F et al. Lactic acidosis during sepsis is related to increased pyruvate production, not deficits in tissue oxygen availability. Ann of Surgery 1996; 224: 97-102.
- Peterson CD. Lactic acidosis. Nutr Clin Pract 2005; 20: 634-45.
- Abramson D, Scalea TM et al. Lactate clearance and survival following injury. J Trauma 1993; 35: 584-88.
- Shapiro NI, Howell MD et al. Serum lactate as a predictor of mortality in emergency department patients with infection. Ann Emerg Med 2005; 45: 524-28.
- Cady LD, Weill MW et al. Quantitation of severity of critical illness with special reference to blood lactate. Crit Care Med 1973; 1: 75-80.
- Trzeciak S. Lac-time? Crit Care Med 2004; 32: 1785-86.
- Nichols JH, Christenson RH et al. Executive summary. The National Academy of Clinical Biochemistry Laboratory Practice Guideline: Evidence-based practice for point of care testing. Clin Chim Acta 2007; 379: 14-28.
- Lavery RF Livingston DH et al. The utility of venous lactate to triage injured patients in the trauma center. J Am Coll Surg 2000; 190: 656-64.
- Wennecke G. Useful tips to avoid preanalytical errors in blood gas testing: metabolites. 2004 Useful tips to avoid preanalytical errors in blood gas testing metabolites.
References
- Mordes J P, Rossini AA. Lactic acidosis. In: Irwin and Rippe’s Intensive Care Medicine (4th edition) Ed: Irwin R, Cera FB, Rippe JM. Philadelphia: Lippincott-Raven, 1999.
- Fall P, Szerlip H. Lactic acidosis: from sour milk to septic shock. J Intensive Care Med 2005; 20: 255-71.
- Yudkin J, Cohen RD. The contribution of the kidney to the removal of lactic acid load under normal and acidotic conditions in the conscious rat. Clin Sci Mol Med 1975; 48: 121-31.
- Cohen RD, Woods HF. Clinical and biochemical aspects of lactic acidosis. Oxford: Blackwell Science.
- Bellomo R. Bench to bedside review: lactate and the kidney. Critical Care 2002; 6: 322-26.
- Sacks DB. Carbohydrates. In: Tietz Textbook of Clinical Chemistry and Molecular Diagnostics (4th edition). St. Louis: Elsevier, 2006.
- Mizcock B. Controversies in lactic acidosis: implications in critically ill patients. JAMA 1987; 258: 497-501.
- Cassaletto J. Differential diagnosis of metabolic acidosis. Emerg Med Clin N Amer. 2005; 23: 771-87.
- Lloyd MH, Iles RA et al. The effect of simulated metabolic acidosis on intracellular pH and lactate metabolism in the isolated perfused rat liver. Clin Sci Mol Med 1973; 45: 543-49.
- Essex DW, Jun DK, Bradley TP. Lactic acidosis secondary to severe anemia in a patient with paroxysmal nocturnal hemoglobinuria. Am J Hematol 1998; 55:110-11.
- Aberman A, Hew E. Lactic acidosis presenting as acute respiratory failure. Am Rev Respir Dis 1978 118: 961-63.
- Foster M, Goodwin S et al. Recurrent life-threatening events and lactic acidosis caused by chronic carbon monoxide poisoning in an infant. Pediatrics 1999; 104: e34-e35.
- Orringer CE, Eustace JC et al. Natural history of lactic acidosis after grand-mal seizures: a model for the study of an anion gap acidosis not associated with hyperkalemia. New Eng J Med 1977; 297: 796-99.
- Freidenburg AS, Brandoff DE et al. Type B lactic acidosis as a severe metabolic complication in lymphoma and leukemia: a case series from a single institution and literature review. Medicine 2007; 86: 225-32.
- Farrell DF Clark AF et al. Absence of pyruvate decarboxylase in man: A cause of congenital lactic acidosis. Science 1975; 187: 1082-84.
- Rallison ML, Meikle AW et al. Hypoglycemia and lactic acidosis associated with fructose-1,6 diphosphatase deficiency. J Pediatrics 1979; 94: 933-36.
- Blanco-Barca O, Gomez-Lado C et al. Pyruvate dehydrogenase deficit associated to the C515T mutation in exon 6 of the E1alpha gene. Rev Neurol 2006; 43: 341-45.
- Stacpoole PW. Metformin and lactic acidosis - guilt by association? Diabetes Care 1998; 21:1587-88.
- John M, Moore CB et al. Chronic hyperlactatemia in HIV-infected patients taking antiretroviral therapy. AIDS 2001; 15: 717-23.
- Bonarek M, Bonnet F et al. Severe lactic acidosis in HIV infected patients treated by nucleoside analog reverse-transcriptase inhibitors – a report of 9 cases. Rev Med Interne 2003; 24: 11-16.
- James JH, Luchette FA et al. Lactate is an unreliable indicator of tissue hypoxia in injury or sepsis. Lancet 1999; 354: 505-08.
- Levraut J, Ciebiera J-P et al. Mild hyperlactatemia in stable septic patients is due to impaired lactate clearance rather than over production. Am J Respir Crit Care Med 1998; 157: 1021-26.
- Vary TC, Siegel JH et al. Effect of sepsis on activity of pyruvate dehydrogenase complex in skeletal muscle and liver. Am J Physiol 1986; 250: E634-E640.
- Gore D, Jahoor F et al. Lactic acidosis during sepsis is related to increased pyruvate production, not deficits in tissue oxygen availability. Ann of Surgery 1996; 224: 97-102.
- Peterson CD. Lactic acidosis. Nutr Clin Pract 2005; 20: 634-45.
- Abramson D, Scalea TM et al. Lactate clearance and survival following injury. J Trauma 1993; 35: 584-88.
- Shapiro NI, Howell MD et al. Serum lactate as a predictor of mortality in emergency department patients with infection. Ann Emerg Med 2005; 45: 524-28.
- Cady LD, Weill MW et al. Quantitation of severity of critical illness with special reference to blood lactate. Crit Care Med 1973; 1: 75-80.
- Trzeciak S. Lac-time? Crit Care Med 2004; 32: 1785-86.
- Nichols JH, Christenson RH et al. Executive summary. The National Academy of Clinical Biochemistry Laboratory Practice Guideline: Evidence-based practice for point of care testing. Clin Chim Acta 2007; 379: 14-28.
- Lavery RF Livingston DH et al. The utility of venous lactate to triage injured patients in the trauma center. J Am Coll Surg 2000; 190: 656-64.
- Wennecke G. Useful tips to avoid preanalytical errors in blood gas testing: metabolites. 2004 Useful tips to avoid preanalytical errors in blood gas testing metabolites.
May contain information that is not supported by performance and intended use claims of Radiometer's products. See also Legal info.


Acute care testing handbook
Get the acute care testing handbook
Your practical guide to critical parameters in acute care testing.
Download now
Related webinar
Evolution of blood gas testing Part 1
Presented by Ellis Jacobs, PhD, Assoc. Professor of Pathology, NYU School of Medicine.
Watch the webinar